


Abstract
The treatment of patients with systemic light chain (AL) amyloidosis is a challenge to hematologists. Despite its generally small size, the underlying clone causes a rapidly progressing, often devastating multiorgan dysfunction through the toxic light chains that form amyloid deposits. Clinical manifestations are deceitful and too often recognized at an irreversible stage. However, hematologists are in the unique position to diagnose AL amyloidosis at a presymptomatic stage, checking biomarkers of amyloid organ involvement in patients with monoclonal gammopathies at higher risk to develop the disease. Adequate technology and expertise are needed for a prompt and correct diagnosis, particularly for ruling out non-AL amyloidoses that are now also treatable. Therapy should be carefully tailored based on severity of organ involvement and clonal characteristics, and early and continual monitoring of response is critical. Three recent randomized clinical trials moved AL amyloidosis to evidence-based era. Above all, the daratumumab-bortezomib combination is a new standard-of-care for newly diagnosed patients, inducing rapid and deep responses that translate into high rates of organ response. The availability of new effective drugs allows to better personalize the therapy, reduce toxicity, and improve outcomes. Patients should be treated within clinical trials whenever possible.
Introduction
Systemic immunoglobulin light chain (AL) amyloidosis is caused by the conversion of light chains (LCs) from their soluble states into highly organized fibrillar aggregates that deposit in tissues, resulting in progressive organ damage and dysfunction.1 Rapid organ deterioration imposes early diagnosis and prompt effective therapies to halt disease progression and possibly rescue organ function.2 LCs, LC aggregates, or preceding intermediaries may induce proteotoxicity, leading to cell dysfunction and death, as shown in cardiac cells.3-5 Cardiomyocytes, in response, produce high levels of natriuretic peptide type-B and its N-terminal fragment (BNP and NT-proBNP, respectively) that are pivotal for patient management.6 Furthermore, amyloid fibrils may cause cell damage and distortion of tissue architecture contributing to organ dysfunction.7 Accordingly, although experimental strategies aiming at reducing amyloid load are being developed, current therapy aims at suppressing the synthesis of the amyloid protein while adapting to patient’s frailty.
Structural features of LCs underlie their amyloidogenicity. Approximately 80% are of the λ isotype, and a limited pool of LC variable region genotypes are responsible for the production of amyloidogenic free LCs (FLC), consistent with a biased selection.8-11 Specific LC germline use can partly explain organ tropism.11,12 Somatic mutations, acquired during clonal selection, determine LC higher flexibility, kinetic instability, and a higher dynamic state that underlie amyloidogenicity and tissue toxicity.13-16 Studies are ongoing to develop LC stabilizers expected to inhibit fibril formation and proteotoxicity.17,18N-glycosylation of κ LCs is more common in patients with AL amyloidosis than in those with monoclonal gammopathy of undetermined significance (MGUS) or multiple myeloma (MM) and can be readily detected in serum by mass spectrometry.19,20 These genetic and posttranslational characteristics may be exploited, possibly through machine learning,21 for detecting patients with plasma cell (PC) dyscrasias at high risk for developing AL amyloidosis, facilitating early diagnosis.22
Several studies have delineated the biologic characteristics of the B-cell/PC clone that is usually small and indolent.23 Amyloidogenic PCs are vulnerable to proteasome inhibitors, accounting for improved clinical outcomes after the introduction of bortezomib.24,25 Cytogenetic abnormalities are detected in approximately 80% of patients via fluorescent in situ hybridization (FISH). t(11;14) is the most frequent (40% to 60% of patients) and is associated with poorer hematologic response rates and overall survival with bortezomib and dexamethasone with or without cyclophosphamide.26 The BCL-2 inhibitor venetoclax is very effective in patients with t(11;14),27 and larger, controlled studies of this agent are warranted. Gain(1q21) is present in 15% to 20% of patients and is associated with poor response to oral melphalan.28 Compared with MGUS/MM, AL amyloidosis shows less intraclonal heterogeneity29 and different transcriptional programs,30 suggesting that AL amyloidosis may be more amenable to eradication than MM.
Case 1: a young, asymptomatic man with MGUS and increased cardiac biomarkers
A 58-year-old, very active man, with low-intermediate risk immunoglobulin G (IgG) λ MGUS diagnosed 1 year earlier, presented at the second yearly control with normal hematology and biochemistry values. Serum monoclonal spike was stable (8 g/L), as well as FLC concentration (λ, 180 mg/L; κ, 20 mg/L; ratio, 0.11), and proteinuria was 220 mg/d with urine immunofixation electrophoresis showing monoclonal λ FLC. Bone marrow biopsy showed 8% λ typic plasma cells, and FISH analysis showed t(11;14) in 35% of interphases. The patient was asymptomatic and working full time. The finding of a significant increase of NT-proBNP to 745 ng/L from the previous 114 ng/L raised the suspicion of heart involvement, and an appropriate workup was started. The abdominal fat aspirate showed amyloid deposits that were typed as AL λ by mass spectrometry. Echocardiography was normal. Cardiac magnetic resonance was consistent with early cardiac amyloidosis. Cardiac troponin I was 0.01 ng/mL, and the patient was classified as cardiac stage31 II. We thoroughly discussed the opportunity to start anticlone therapy, carefully considering toxicity and benefit balance. The patient decided to start therapy, and because he was eligible for autologous stem cell transplantation (ASCT), we started treatment with cyclophosphamide, bortezomib, and dexamethasone (CyBorD). During the first course, asymptomatic orthostatic hypotension (80/50 mm Hg) occurred but resolved spontaneously. After 4 courses, a very good partial response (VGPR) was achieved (λ, 48 mg/L; κ, 17 mg/L; difference between involved and uninvolved FLC [dFLC], 31 mg/L; positive serum and urine immunofixation). Hemopoietic stem cells were harvested uneventfully. However, 3 months later, NT-proBNP increased to 954 ng/L, and the patient reported unusual fatigue after strenuous physical exercise while maintaining VGPR. The patient consented to proceed to high-dose melphalan (200 mg/m2). The procedure was uneventful. Three months after ASCT, he achieved complete hematologic response (CR), with reduction of NT-proBNP to 584 ng/L, thus qualifying for cardiac response. Eighteen months after ASCT, the patient is in CR, with negative minimal residual disease (MRD) by next-generation flow cytometry, and NT-proBNP is now 98 ng/L.
Comments about patient 1
This patient illustrates the possibility to diagnose AL amyloidosis at a very early asymptomatic stage and to fully exploit the therapeutic armamentarium and restore organ function. About 3% to 5% of patients with a known precursor PC disorder will progress to AL amyloidosis. However, even in referral centers, it takes a median time of almost 1 year to reach the diagnosis, based on signs or symptoms, in patients with preexisting PC dyscrasias.32 When symptoms manifest, even a minor delay translates into significant shortening of survival.33 For this reason, we advocated the use of sensitive markers of amyloid organ involvement during the follow-up of individuals with MGUS to facilitate a presymptomatic diagnosis.34 In this patient, an increase of NT-proBNP raised suspicion of amyloid cardiac involvement. NT-proBNP is a very sensitive marker, but it is not specific (being elevated in other conditions), and heart involvement can be confirmed by echocardiography or magnetic resonance. Our diagnostic approach is reported in Figure 1. Once diagnosis is established, it is necessary to assess the risk to carefully balance expected benefit and possible treatment toxicity. Cardiac involvement is the main determinant of frailty and survival. Current validated staging systems are reported in Figure 2.31,35-39 The patient was cardiac stage II and considering the young age and perfect fit, he was eligible for high-dose melphalan and ASCT. Eligibility criteria vary for centers (Figure 3); however, only ∼20% of newly diagnosed patients are eligible for this intensive treatment. ASCT is safe, with <3% treatment-related mortality in referral centers, and highly effective, providing ≥VGPR in approximately 70% of patients and a median survival > 15 years in patients achieving CR.40,41 Induction therapy with bortezomib-based regimens should be considered in all transplant-eligible patients.42,43 Furthermore, the recently concluded Andromeda trial, reported an unprecedented high rate of deep hematologic responses (≥VGPR in 78.5%) for daratumumab plus CyBorD, which was recently approved by the US Food and Drug Administration and European Medicines Agency for newly diagnosed AL amyloidosis.44 Daratumumab-CyBorD is a new standard of care and will become also the preferred induction therapy before ASCT. Importantly, daratumumab-CyBorD is also effective in patients whose amyloid clones harbor the t(11;14) who have poorer outcomes with CyBorD alone.44 Because daratumumab was not available in Italy at the time this patient was seen, he received CyBorD achieving VGPR after 4 courses. Based on data suggesting that ASCT can be deferred to relapse after successful induction, without detrimental effects on overall survival, we elected to postpone ASCT.45-47 This practice is common in Europe. Actually, in a survey including 3064 patients treated between 2011 and 2018 in 10 European referral centers, ASCT was used as frontline therapy in 6% of patients and as second-line therapy in 10%.48 Although some centers prefer transplant for all eligible patients upfront, we generally prefer to defer ASCT in subjects who attain CR and/or organ response after induction. However, at our center, we proceed to ASCT in patients with MM. In this patient, the appearance of mild symptoms and increase in NT-proBNP suggested that attaining VGPR was not sufficient to halt disease progression, and the patient proceeded to ASCT. Approximately 30% of patients do not respond to first-line treatment, and they should promptly be switched to another class of drugs. Quality of organ response is strictly dependent on the depth of hematology response, and although in some patients VGPR or even PR is enough to reach it, in patients with highly toxic LCs, persistence of even MRD may be sufficient to hamper amelioration of organ function.49 In patients who achieve CR without organ response, MRD evaluation may help decide regarding further therapy. Proposed criteria for grading organ response are now being validated.50 The criteria for hematologic and organ response are reported in Table 1.39,51-54 In this fit patient, stem cell collection was uneventful, although the incidence of major complications (hypotension, hypoxia, cardiac arrhythmia, and fluid retention) during stem cell mobilization and collection is approximately 15%. The patient was eligible for full-dose melphalan (200 mg/m2), which is important to optimize outcomes, and reached CR and cardiac response.
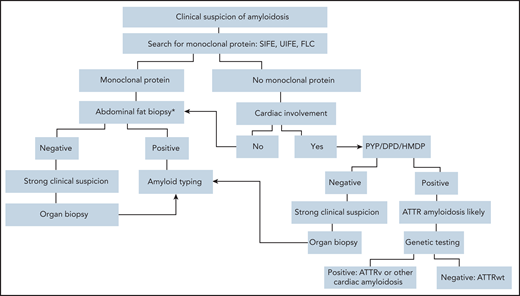
Diagnostic algorithm in symptomatic patients. Ideally, AL amyloidosis should be diagnosed at a presymptomatic stage. When amyloidosis is suspected, the first step should be searching for a monoclonal protein with sensitive technology. Patients with suspect cardiac involvement in whom monoclonal components are not detected may undergo a biopsy-free diagnostic process.58 Scintigraphy with bone-seeking tracers, such as DPD, PYP, and HMDP, can diagnose ATTR amyloidosis without a biopsy in the absence of a plasma cell dyscrasia, but cardiac uptake can be seen also in patients with AL amyloidosis. In the presence of a monoclonal component biopsy-based diagnosis and typing with mass spectrometry,102 or, in highly specialized laboratories, with immunohistochemistry or immuno-electron microscopy102 are mandatory. Amyloid deposits can be demonstrated with minimally invasive procedures, such as biopsy of abdominal fat, bone marrow, and minor salivary glands. Abdominal fat aspirate has excellent feasibility and good sensitivity (∼85% when associated with bone marrow biopsy) in AL amyloidosis.103 If less invasive biopsies are negative, a biopsy of an affected organ will typically be required. Genetic testing allows discrimination between hereditary and wild-type TTR amyloidosis and identification of rarer hereditary variants. *In patients with a monoclonal protein, amyloid deposits should be searched also in the bone marrow biopsy. DPD, scintigraphy with 99mTc 3,3-diphosphono1,2-propanodicarboxylic acid; FLC, circulating free light chains; HMDP, scintigraphy with 99mTc-hydroxymethylene diphosphonate; PYP, scintigraphy with 99mTc-pyrophosphate; SIFE, serum immunofixation electrophoresis; UIFE, urine immunofixation electrophoresis.
Diagnostic algorithm in symptomatic patients. Ideally, AL amyloidosis should be diagnosed at a presymptomatic stage. When amyloidosis is suspected, the first step should be searching for a monoclonal protein with sensitive technology. Patients with suspect cardiac involvement in whom monoclonal components are not detected may undergo a biopsy-free diagnostic process.58 Scintigraphy with bone-seeking tracers, such as DPD, PYP, and HMDP, can diagnose ATTR amyloidosis without a biopsy in the absence of a plasma cell dyscrasia, but cardiac uptake can be seen also in patients with AL amyloidosis. In the presence of a monoclonal component biopsy-based diagnosis and typing with mass spectrometry,102 or, in highly specialized laboratories, with immunohistochemistry or immuno-electron microscopy102 are mandatory. Amyloid deposits can be demonstrated with minimally invasive procedures, such as biopsy of abdominal fat, bone marrow, and minor salivary glands. Abdominal fat aspirate has excellent feasibility and good sensitivity (∼85% when associated with bone marrow biopsy) in AL amyloidosis.103 If less invasive biopsies are negative, a biopsy of an affected organ will typically be required. Genetic testing allows discrimination between hereditary and wild-type TTR amyloidosis and identification of rarer hereditary variants. *In patients with a monoclonal protein, amyloid deposits should be searched also in the bone marrow biopsy. DPD, scintigraphy with 99mTc 3,3-diphosphono1,2-propanodicarboxylic acid; FLC, circulating free light chains; HMDP, scintigraphy with 99mTc-hydroxymethylene diphosphonate; PYP, scintigraphy with 99mTc-pyrophosphate; SIFE, serum immunofixation electrophoresis; UIFE, urine immunofixation electrophoresis.
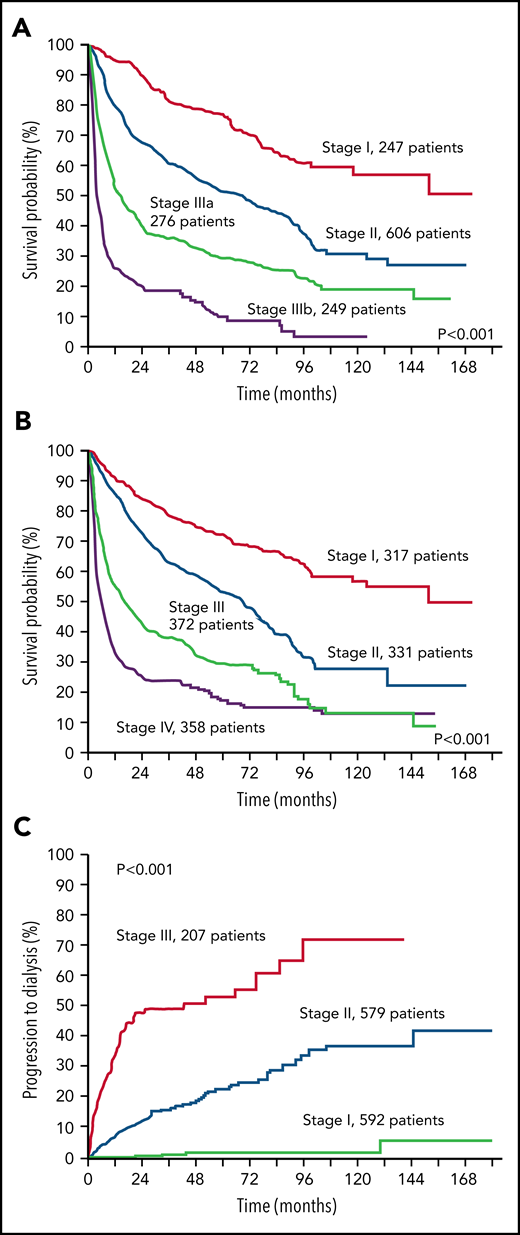
Stratification of 1378 patients with AL amyloidosis according to the validated staging systems. (A) European modification of the Mayo Clinic 2004 staging system.31,35 The staging system is based on NT-proBNP and cardiac troponin (cTn). Troponin I was used in this figure. Cutoffs are 332 ng/L for NT-proBNP and 100, 35, and 54 ng/L for cTnI, cTnT, and high-sensitivity cTnT, respectively. B-type natriuretic peptide can substitute for NT-proBNP in the staging system (cutoff, 81 ng/L).36 Patients with stage I, II, and III have 0, 1, and 2 markers above the cutoff, respectively. Patients with stage III are classified as stage IIIa or IIIb according to concentration of NT-proBNP below or above 8500 ng/L. (B) Revised Mayo Clinic staging system.37 The staging system is based on NT-proBNP, cTn, and dFLC. Cutoffs are 1800 ng/L for NT-proBNP and 25 ng/L for cTnT. Troponin I can substitute for cTnT (cutoff, 70 ng/L)38 and was used in this figure. Patients with stage I, II, III, and IV have 0, 1, 2, and 3 markers above the cutoff, respectively. (C) Renal staging system.39 The staging system is based on estimated glomerular filtration rate (eGFR) and proteinuria. Cutoffs are 50 mL/min per 1.73 m2 for eGFR and 5 g/24 h for proteinuria. Patients with stage I, II, and III have 0, 1, and 2 markers above the cutoff, respectively.
Stratification of 1378 patients with AL amyloidosis according to the validated staging systems. (A) European modification of the Mayo Clinic 2004 staging system.31,35 The staging system is based on NT-proBNP and cardiac troponin (cTn). Troponin I was used in this figure. Cutoffs are 332 ng/L for NT-proBNP and 100, 35, and 54 ng/L for cTnI, cTnT, and high-sensitivity cTnT, respectively. B-type natriuretic peptide can substitute for NT-proBNP in the staging system (cutoff, 81 ng/L).36 Patients with stage I, II, and III have 0, 1, and 2 markers above the cutoff, respectively. Patients with stage III are classified as stage IIIa or IIIb according to concentration of NT-proBNP below or above 8500 ng/L. (B) Revised Mayo Clinic staging system.37 The staging system is based on NT-proBNP, cTn, and dFLC. Cutoffs are 1800 ng/L for NT-proBNP and 25 ng/L for cTnT. Troponin I can substitute for cTnT (cutoff, 70 ng/L)38 and was used in this figure. Patients with stage I, II, III, and IV have 0, 1, 2, and 3 markers above the cutoff, respectively. (C) Renal staging system.39 The staging system is based on estimated glomerular filtration rate (eGFR) and proteinuria. Cutoffs are 50 mL/min per 1.73 m2 for eGFR and 5 g/24 h for proteinuria. Patients with stage I, II, and III have 0, 1, and 2 markers above the cutoff, respectively.
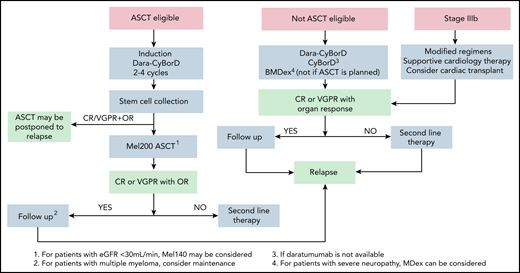
Upfront treatment algorithm for AL amyloidosis. At our center, the first step in the design of the therapeutic strategy is assessing eligibility for ASCT. However, the role of ASCT in AL amyloidosis is not supported by controlled trials and is challenged by the availability of very effective treatments like Dara-CyBorD. Yet, large long-term outcome studies, which are lacking for newer combinations, show that ASCT grants long-lasting responses with a prolonged improvement in survival.40 At our center, eligibility for stem cell transplant with full dose (200 mg/m2) melphalan requires the following: age <70 years, Eastern Cooperative Oncology Group performance status <2, NT-proBNP <5000 ng/L, troponin T <60 ng/L, left ventricular ejection fraction > 45%, New York Heart Association class <III, systolic blood pressure ≥100 mm Hg, glomerular filtration rate >50 mL/min unless on dialysis, bilirubin <2 mg/dL, and diffusing capacity of the lungs for carbon monoxide >50%. At some referral centers, including ours, transplant is performed only in patients who are eligible for full-dose melphalan.42 Eligibility can be extended with a risk-adapted conditioning dose of melphalan (100-140 mg/m2).104 However, there is no evidence that reduced-dose melphalan is better than bortezomib-based chemotherapy, and this approach is even more challenged by newer powerful nontransplant regimens.44,105 Induction should be performed with Dara-CyBorD. If daratumumab is not yet available, CyBorD can be used. In subjects who have contraindications to bortezomib (eg, peripheral neuropathy), daratumumab-based induction without bortezomib may be considered, or ASCT can be performed upfront. In some cases, a satisfactory response (CR or VGPR, accompanied by organ response) can be achieved after induction alone. In these cases, treatment can be discontinued, and close monitoring initiated to detect hematologic relapse before this causes progression of organ involvement. This sequential approach reduces treatment-related mortality to less than 1% and allows fully exploiting modern powerful regimens in transplant-eligible patients.45-47 This approach is adopted at some referral centers, but it is not supported by controlled studies. If patients achieve less than a satisfactory response after ASCT, consolidation with bortezomib may improve the outcome.106 Dara-CyBorD is a new standard of care for patients who do not undergo ASCT. In patients who are potentially eligible for transplantation, melphalan should be avoided. In elderly individuals, BMDex may be considered. Patients with contraindications to bortezomib can be treated with MDex. Patients with advanced cardiac involvement (stage IIIb) should start bortezomib based treatment immediately, with attenuated dosages and under close medical control. Cardiac transplant may be considered. Dara-CyBorD, daratumumab, cyclophosphamide, bortezomib, and dexamethasone; OR, organ response.
Upfront treatment algorithm for AL amyloidosis. At our center, the first step in the design of the therapeutic strategy is assessing eligibility for ASCT. However, the role of ASCT in AL amyloidosis is not supported by controlled trials and is challenged by the availability of very effective treatments like Dara-CyBorD. Yet, large long-term outcome studies, which are lacking for newer combinations, show that ASCT grants long-lasting responses with a prolonged improvement in survival.40 At our center, eligibility for stem cell transplant with full dose (200 mg/m2) melphalan requires the following: age <70 years, Eastern Cooperative Oncology Group performance status <2, NT-proBNP <5000 ng/L, troponin T <60 ng/L, left ventricular ejection fraction > 45%, New York Heart Association class <III, systolic blood pressure ≥100 mm Hg, glomerular filtration rate >50 mL/min unless on dialysis, bilirubin <2 mg/dL, and diffusing capacity of the lungs for carbon monoxide >50%. At some referral centers, including ours, transplant is performed only in patients who are eligible for full-dose melphalan.42 Eligibility can be extended with a risk-adapted conditioning dose of melphalan (100-140 mg/m2).104 However, there is no evidence that reduced-dose melphalan is better than bortezomib-based chemotherapy, and this approach is even more challenged by newer powerful nontransplant regimens.44,105 Induction should be performed with Dara-CyBorD. If daratumumab is not yet available, CyBorD can be used. In subjects who have contraindications to bortezomib (eg, peripheral neuropathy), daratumumab-based induction without bortezomib may be considered, or ASCT can be performed upfront. In some cases, a satisfactory response (CR or VGPR, accompanied by organ response) can be achieved after induction alone. In these cases, treatment can be discontinued, and close monitoring initiated to detect hematologic relapse before this causes progression of organ involvement. This sequential approach reduces treatment-related mortality to less than 1% and allows fully exploiting modern powerful regimens in transplant-eligible patients.45-47 This approach is adopted at some referral centers, but it is not supported by controlled studies. If patients achieve less than a satisfactory response after ASCT, consolidation with bortezomib may improve the outcome.106 Dara-CyBorD is a new standard of care for patients who do not undergo ASCT. In patients who are potentially eligible for transplantation, melphalan should be avoided. In elderly individuals, BMDex may be considered. Patients with contraindications to bortezomib can be treated with MDex. Patients with advanced cardiac involvement (stage IIIb) should start bortezomib based treatment immediately, with attenuated dosages and under close medical control. Cardiac transplant may be considered. Dara-CyBorD, daratumumab, cyclophosphamide, bortezomib, and dexamethasone; OR, organ response.
Criteria for hematologic and organ response
| Hematologic response | Criteria |
|---|---|
| Complete response (CR) | Both criteria must be met:
|
| Very good partial response (VGPR) | dFLC <40 mg/L |
| Partial response (PR) | dFLC decrease >50% |
| No response (NR) | Less than a partial response |
| Organ response | |
| Cardiac response | Decrease of NT-proBNP by >30% and 300 ng/L (if baseline NT-proBNP >650 ng/L) |
| Renal response | At least 30% decrease in proteinuria or drop below 0.5 g/24 h, in the absence of renal progression defined as a >25% decrease in eGFR |
| Hepatic response | 50% decrease in abnormal alkaline phosphatase value or decrease in radiographic liver size by ≥2 cm |
| Hematologic response | Criteria |
|---|---|
| Complete response (CR) | Both criteria must be met:
|
| Very good partial response (VGPR) | dFLC <40 mg/L |
| Partial response (PR) | dFLC decrease >50% |
| No response (NR) | Less than a partial response |
| Organ response | |
| Cardiac response | Decrease of NT-proBNP by >30% and 300 ng/L (if baseline NT-proBNP >650 ng/L) |
| Renal response | At least 30% decrease in proteinuria or drop below 0.5 g/24 h, in the absence of renal progression defined as a >25% decrease in eGFR |
| Hepatic response | 50% decrease in abnormal alkaline phosphatase value or decrease in radiographic liver size by ≥2 cm |
Hematologic, cardiac, and renal response criteria have been validated,39,51 whereas hepatic response criteria have been obtained by consensus.52 A value adequate to measure hematologic response is deemed to be 50 mg/L. For patients with dFLC between 50 and 20 mg/L, hematologic response other than complete response (ungraded) is reached when dFLC falls below 10 mg/L.53,54
Case 2: a woman with rapidly worsening dyspnea and peripheral edema
A 58-year-old woman began to experience exertional dyspnea. Three months later, she was hospitalized because worsening dyspnea and pleural effusion. Echocardiogram reported left ventricular hypertrophy with preserved ejection fraction (55%). Diuresis resolved pleural effusion, and she was discharged. Over the next 6 months, symptoms worsened, and cardiac catheterization showed normal coronaries. Two months later, the patient was seen in another cardiology center because of recurrent cardiac syncope and nonsustained ventricular tachycardia. An implantable cardioverter defibrillator (ICD) was implanted, and oral amiodarone was started. Echocardiography showed diastolic dysfunction and increased wall thickness (posterior wall, 16 mm; interventricular septum, 15 mm) with reduced ejection fraction (43%). The suspicion of cardiac amyloidosis was raised, and a scintigraphy with 99mTc 3,3-diphosphono1,2-propanodicarboxylic acid and an endomyocardial biopsy were performed. The scintigraphy showed no cardiac uptake, and the biopsy showed amyloid deposits that were not further characterized. NT-proBNP was 10 740 ng/L, and troponin I was 0.44 ng/mL. The patient was referred to our center 2 months later, more than 1 year after the onset of symptoms. She reported fatigue, dyspnea for minor exertion, petechiae, and ecchymoses in the upper part of the body. Physical examination revealed mild macroglossia, jugular vein distention, pleural effusion, hepatomegaly, distal edema, and hypotension (86/53 mm Hg). Serum and urine electrophoresis and immunofixation revealed an IgG κ spike (28 g/L) with free κ LCs and Bence Jones proteinuria. Serum κ FLCs were 303 mg/L (ratio, 25.2; dFLC, 291 mg/L). A bone marrow biopsy speciman showed 31% monotypic κ PC with gain1q21 and amyloid deposits. No other myeloma-defining events were present. Abdominal fat aspirate was positive for amyloid that was typed as AL κ by immuno-electron microscopy. NT-proBNP was 15 585 ng/L, troponin I was 0.53 ng/mL, and echocardiography confirmed advanced amyloid involvement. The patient was stage IIIb, and because there was no sign of other organ damage, we started evaluation for cardiac transplantation. In the meantime, she started treatment with CyBorD with reduced bortezomib (1 mg/m2) and dexamethasone (20 mg) dosage. After the second cycle, she experienced worsening of heart failure that required hospitalization. Investigations revealed reduction of the serum monoclonal component to 19 g/L and of dFLC to 35 mg/L, qualifying for VGPR, whereas NT-proBNP increased to 16 834 ng/L and troponin I to 0.63 ng/L. Three days after admission, the patient collapsed and died with pulseless electrical activity.
Comments about patient 2
This case illustrates the devastating effects of delayed diagnosis: in this rapidly progressing disease, “time is life,” and even few months can irreversibly compromise any chance to benefit from effective therapies. Symptoms that should trigger an appropriate workup for amyloidosis are listed in Table 2. The diagnosis of cardiac amyloidosis can be augmented by machine learning applied to electrocardiographic and echocardiographic data.55 In the European survey, the percentage of patients presenting with very advanced cardiac damage (stage IIIb) remained the same (15%-16%), and median survival remained disappointingly low (4.5 months) in the periods 2004 to 2010 and 2011 to 2018.56 A recent survey among US community and academic hematologists showed that low disease awareness, inadequate referral practices, and inadequate screening/testing procedures account for a 1- to 1.5-year diagnostic delay.57 Scintigraphy with bone tracers (PYP or DPD) is used to search for cardiac transthyretin amyloidosis that usually shows strong uptake.58 However, the presence of the monoclonal protein should be ascertained first, using appropriate methods (Figure 1), because its presence makes cardiac biopsy mandatory and scintigraphy unnecessary. In this case, the cardiologists elected to perform scintigraphy and cardiac biopsy concomitantly. Because of recurrent syncope and registered ventricular arrhythmias, an ICD was implanted. Probably, patients with very advanced cardiac amyloidosis (New York Heart Association class 4, NT-proBNP >8500 ng/L, and systolic blood pressure <90 mm Hg) are not appropriate candidates for ICD implantation.59 The patient reported recurrent purpura, a common finding particularly in advanced stages of the disease, that is attributed to amyloid infiltration of blood vessels. Coagulation abnormalities are multifactorial, including factor x and other factor deficiencies, and occur in a significant proportion of patients.60 These extremely fragile patients are very sensitive to treatment toxicity, and we attenuated treatment dosages. Although early and deep response is associated with improved survival in patients with stage IIIb,61 in this subject, cardiac involvement was likely already too advanced. At present, there is no treatment capable of improving the outcome of most patients with stage IIIb. However, subcutaneous daratumumab can induce rapid and deep responses, which makes it a valuable option in these patients. A multicenter phase 2 trial of single-agent daratumumab in subjects with stage IIIb is underway (NCT04131309). Moreover, removing amyloid deposits with antibodies is expected to improve outcomes and is currently being tested in clinical trials (Table 3). Young patients with stage IIIb with isolated heart involvement should be considered for cardiac transplantation that is now associated with outcomes comparable to nonamyloid cardiomyopathy.62 However, scarcity of donors frequently renders time on waiting list incompatible with patient’s needs.
Presymptomatic hints, symptoms, and signs leading to the diagnosis of AL amyloidosis
| Presymptomatic | Symptomatic | ||
|---|---|---|---|
| Organ involved | Symptoms | Signs | |
Increased markers of possible amyloid organ involvement:
| Heart | Reduced exercise tolerance Dyspnea at rest or exertion Fatigue Syncope Angina | Lower extremity edema Pleural effusions Jugular vein distension Arrhythmia Thickened ventricular walls and low voltages on ECG |
| Kidney | Loss of appetite Fatigue and weakness | Lower extremity edema Anasarca | |
| Soft tissues | Jaw or buttock claudication Carpal tunnel (often bilateral) Dysarthria | Periorbital (upper body) purpura Macroglossia Nail dystrophy Shoulder pad Arthropathy Myopathy | |
| Peripheral nervous system including autonomic nervous system | Length dependent sensory-motor neuropathy Lipothymia Taste alterations Early satiety | Orthostatic hypotension Intestinal dysmotility Erectile dysfunction Voiding dysfunction | |
| Liver | Right upper quadrant tenderness Jaundice Early satiety | Hepatomegaly Weight loss | |
| Gastrointestinal tract | Abdominal discomfort | Malabsorption Gastrointestinal bleeding Diarrhea Weight loss | |
| Presymptomatic | Symptomatic | ||
|---|---|---|---|
| Organ involved | Symptoms | Signs | |
Increased markers of possible amyloid organ involvement:
| Heart | Reduced exercise tolerance Dyspnea at rest or exertion Fatigue Syncope Angina | Lower extremity edema Pleural effusions Jugular vein distension Arrhythmia Thickened ventricular walls and low voltages on ECG |
| Kidney | Loss of appetite Fatigue and weakness | Lower extremity edema Anasarca | |
| Soft tissues | Jaw or buttock claudication Carpal tunnel (often bilateral) Dysarthria | Periorbital (upper body) purpura Macroglossia Nail dystrophy Shoulder pad Arthropathy Myopathy | |
| Peripheral nervous system including autonomic nervous system | Length dependent sensory-motor neuropathy Lipothymia Taste alterations Early satiety | Orthostatic hypotension Intestinal dysmotility Erectile dysfunction Voiding dysfunction | |
| Liver | Right upper quadrant tenderness Jaundice Early satiety | Hepatomegaly Weight loss | |
| Gastrointestinal tract | Abdominal discomfort | Malabsorption Gastrointestinal bleeding Diarrhea Weight loss | |
Antiamyloid therapies currently under development for AL amyloidosis
| Agent | Target | Comments |
|---|---|---|
| CAEL-101 | Amyloid fibrils |
|
| Birtamimab | Amyloid fibrils |
|
| Doxycycline | Amyloid deposits Amyloid light chains |
|
| Agent | Target | Comments |
|---|---|---|
| CAEL-101 | Amyloid fibrils |
|
| Birtamimab | Amyloid fibrils |
|
| Doxycycline | Amyloid deposits Amyloid light chains |
|
Case 3: a man with rheumatoid arthritis, proteinuria, smoldering MM, and bilateral carpal tunnel syndrome
A 70-year-old man with a 1-year history of rheumatoid arthritis treated with hydroxychloroquine and prednisone (2.5 mg/d) and of smoldering MM (IgG λ; FLC ratio, 0.08; dFLC, 141 mg/L; bone marrow PC infiltrate, 25%; t(11;14) in 25% of interphases) was referred to our center because of bilateral carpal tunnel syndrome and proteinuria of 1.2 g/d (creatinine, 0.71 mg/dL). There was no sign of amyloid cardiac involvement (normal echocardiography; NT-proBNP, 42 ng/L; troponin I, 0.03 ng/mL). Serum amyloid A apolipoprotein (SAA) level was normal. Abdominal fat aspirate was positive, and the amyloid deposits reacted with anti-λ (and not with anti-SAA and anti-transthyretin) antibodies at immuno-electron microscopy. A diagnosis of AL amyloidosis with renal and soft tissue involvement was made, and the patient was enrolled in the ANDROMEDA trial and randomized to the daratumumab-CyBorD arm. After 2 infusions, treatment was interrupted because of a respiratory infection. However, by then, a VGPR had been reached (dFLC, 13 mg/L; free light chain ratio [FLCR], 0.61 ; positive serum immunofixation) with renal response (proteinuria, 0.5 g/d). Cycle 2 was also interrupted after the second infusion because of a respiratory infection. The following 4 cycles were completed without complications, but we elected to withhold daratumumab maintenance. The patient is currently asymptomatic 2 years after treatment discontinuation, and VGPR is maintained (dFLC, 14 mg/L; FLCR, 0.81; positive serum immunofixation).
Comments on patient 3
In this patient, symptoms were recognized early, and the diagnosis was made when he was still renal stage I. However, this subject had a chronic inflammation (rheumatoid arthritis) that can give rise to AA (reactive) amyloidosis. Amyloidosis reactive to chronic phlogosis is caused by deposition of SAA-derived amyloid fibrils and presents with renal damage.63 Treatment is aimed at controlling the underlying inflammation. Moreover, carpal tunnel syndrome can be found both in AL and in transthyretin amyloidosis. Thus, accurate tissue typing was mandatory.
In this patient, bone marrow PC infiltrate was higher than 20%. This measure of clonal bulk is an additional negative prognostic factor independent of cardiac involvement,64 as well as dFLC level.37,53,54 The patient could be treated with daratumumab-CyBorD in the ANDROMEDA trial. This regimen is well tolerated, but 1 of the most common adverse events with daratumumab in AL amyloidosis is respiratory infection,65 as occurred in this subject who was also immunocompromised by steroid treatment of rheumatoid arthritis. Nevertheless, the infection was manageable, and VGPR was reached after only 2 infusions. Prophylaxis of infections may be useful in patients with AL amyloidosis treated with daratumumab as was shown in MM.66 Besides its high efficacy, the ability of daratumumab to induce deep responses after the first infusion67,68 is particularly relevant in patients with AL amyloidosis who need to reach a deep reduction of the amyloid LCs as soon as possible.
The role of maintenance has not been adequately explored in patients with AL amyloidosis. In the ANDROMEDA study, treatment with daratumumab could be continued for up to 2 years if well tolerated, but there was no control arm for maintenance. In our patient, who was prone to infections, we elected to withhold maintenance. Controlled studies are warranted to ascertain whether maintenance has a role in AL amyloidosis. At our center and at the Mayo Clinic,69 maintenance is considered in patients with large clones (eg, fulfilling the SLiM CRAB criteria) and/or high-risk cytogenetics.
Case 4: an older man with macroglossia, peripheral edema, and dysautonomia
A 72-year-old man developed worsening speech difficulty caused by slowly progressing macroglossia. After 18 months, foamy urine, peripheral edema, impotence, and symptomatic postural hypotension with occasional syncope ensued, and the patient sought medical advice. Proteinuria was present and the general practitioner suspected amyloidosis and referred the patient to our center. He presented with typical macroglossia and submandibular gland swelling, nephrotic syndrome (proteinuria, 5.9 g/d; normal creatinine), symptomatic orthostatic hypotension, and hepatomegaly (8 cm below costal margin). Echocardiography revealed amyloid heart involvement, NT-proBNP was 5500 ng/L, and cardiac troponin I was 0.05 ng/L. Alkaline phosphatase was elevated (480 U/L; upper reference limit, 150 U/L) with normal bilirubin and aminotransferases. Immunofixation electrophoresis showed monoclonal λ FLC in serum and urine, and circulating λ FLC concentration was 849 mg/L (ratio, 85; dFLC, 840 mg/L). Bone marrow plasma cell infiltrate was 16%, with no chromosomal abnormalities by interphase fluorescence in situ hybridization (iFISH). Abdominal fat aspirate showed amyloid deposits characterized as AL-λ type by immuno-electron microscopy. A diagnosis of AL amyloidosis with cardiac, renal, liver, soft tissue, and autonomic nervous system involvement was made, and the patient was enrolled in a clinical trial comparing melphalan and dexamethasone (MDex) with MDex plus bortezomib (BMDex). The patient was randomized to the BMDex arm. The use of fitted elastic leotards was recommended. Diuretic therapy was initiated before chemotherapy with furosemide 25 mg/d, and the patient lost 2 kg in 1 week. Higher doses of furosemide were not tolerated because of worsening hypotension. During the first BMDex course, fluid retention worsened again. By the end of cycle 1, fluid retention and hypotension resolved and did not recur during subsequent cycles. After 3 cycles, a partial response was reached, and VGPR was attained after 6 cycles and maintained at the completion of cycle 8 (positive urine immunofixation; dFLC, 33 mg/L; ratio, 0.30), with cardiac (NT-proBNP 686 ng/L), renal (proteinuria 1.4 g/d), and liver (alkaline phosphatase 150 U/L) response, and treatment was discontinued. The patient was followed every 6 months for 5 years, when an increase in dFLC was observed (141 mg/L; ratio, 0.06) with stable NT-proBNP (767 ng/L) and proteinuria (1.6 g/d). The only symptoms were mild fatigue and persistent involvement of soft tissues. Rescue treatment with ixazomib, lenalidomide, and dexamethasone was started. After 2 cycles, partial hematologic response was achieved (dFLC, 61 mg/L; ratio, 0.16) with stable symptoms, increased NT-proBNP (3835 ng/L), stable proteinuria (1.4 g/d), and an increase in serum creatinine (from 0.77 to 1.4 mg/dL). Treatment was continued for 4 more cycles with stable hematologic and organ parameters. After lenalidomide discontinuation, NT-proBNP level decreased to 480 ng/L. Eighteen months later, a new rise in dFLC was documented (169 mg/L; ratio, 0.09) that was associated with stable NT-proBNP (576 ng/L), proteinuria (1.5 g/d), and creatinine (1.5 mg/dL). Rescue treatment with pomalidomide and dexamethasone was started. A VGPR was re-established after 3 cycles (dFLC, 21 mg/L; ratio, 0.48; persistent positive urine immunofixation). A suspected pomalidomide-related increase in NT-proBNP was observed during therapy (up to 3057 ng/L) while renal involvement remained stable (proteinuria, 1.3 g/d; creatinine, 1.3 mg/dL). The patient is now 81 years old, and he is enjoying an active life despite persistent macroglossia, with resolution of symptoms of heart and renal involvement and of hypotension.
Comments on patient 4
Symptoms of AL amyloidosis depend on organ involvement and can be often mistaken for manifestations of more common diseases. However, typical soft tissue involvement and combination of signs of simultaneous involvement of different organs can be a clue to diagnosis as in this patient. Yet, multiorgan involvement makes these patients frail and difficult to treat intensively. Supportive measures to manage fluid retention, hypotension, and malnutrition should be started early, possibly before initiation of chemotherapy (Table 4).70 Dexamethasone can exacerbate fluid retention and arrhythmias and should be used at lower dosage (≤20 mg) in patients with advanced heart involvement (New York Heart Association class III or IV, cardiac stage IIIb, repetitive ventricular arrhythmias).71 At diagnosis, this patient was cardiac stage II and renal stage II, and he was not a candidate for stem cell transplantation because of age, heart involvement, and hypotension, qualifying as intermediate risk. Bortezomib is the most used agent in upfront therapy, based on retrospective uncontrolled studies.35,72 Running controlled clinical trials has been difficult in AL amyloidosis because of the relative rarity of this condition and the easy access to drugs licensed for MM. This patient was enrolled in the AC-004-EU/EMN-03 trial comparing MDex and BMDex (NCT01277016). This academic, investigator-initiated study showed that BMDex grants higher hematologic response rates than MDex (81% vs 57%; VGPR/CR, 64% vs 39%) and was the only study to demonstrate an overall survival advantage for the experimental arm in AL amyloidosis.73 BMDex is well tolerated and relatively inexpensive. Moreover, it can potentially overcome the effect of the 2 most common chromosomal abnormalities found in this disease: t(11;14) and gain 1q(21).26,28 In this patient, BMDex induced VGPR with cardiac, renal, and liver response that lasted 5 years. It has recently been shown that restaging discriminates survival even after upfront therapy.74 The patient had asymptomatic hematologic relapse and was rescued when he still was cardiac stage II. There are no validated criteria for hematologic progression in AL amyloidosis, and there is disagreement on when treatment should be started at relapse.75-77 This is relevant not only for individual patient management but also for the lack of validated definition of progression-free survival in clinical trials. However, it is generally agreed that organ progression should not be awaited, because this is associated with shorter survival and time to dialysis.39,51 Indeed, it is common practice at different referral centers to start treatment when early, relatively small increases in FLC (approximately 40% of baseline value) are detected.78,79 Small increases in FLC should be regarded cautiously particularly in patients who presented with advanced cardiac involvement. Rescue therapy is discussed in Figure 4. This patient was rescued with a combination of ixazomib and lenalidomide,80 and pomalidomide at first and second relapse, before progression became symptomatic. Although he never reached CR, his disease was controlled for 10 years, with a good quality of life.
Supportive therapy in AL amyloidosis
| Condition | Supportive measures |
|---|---|
| Fluid retention |
|
| Hypotension |
|
| Neuropathy |
|
| Diarrhea |
|
| Malnutrition |
|
| End-stage renal disease |
|
| Heart failure |
|
| Condition | Supportive measures |
|---|---|
| Fluid retention |
|
| Hypotension |
|
| Neuropathy |
|
| Diarrhea |
|
| Malnutrition |
|
| End-stage renal disease |
|
| Heart failure |
|
See Cibeira et all.70
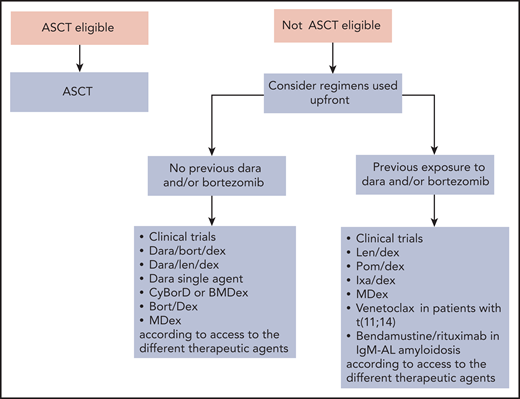
Rescue treatment algorithm for AL amyloidosis. Relapsed/refractory patients should be enrolled in clinical trials whenever possible. Treating relapsed patients with a different class of agents than that used in upfront therapy is associated with prolonged PFS but has no impact on OS.107 Thus, if response to the previous line of therapy lasted at least 12 to 18 months, retreatment with the same drugs can be considered. Eligible patients who did not perform ASCT can be transplanted at relapse. At present, many relapsing patients have not been exposed to daratumumab, and this agent alone or combined with bortezomib or lenalidomide can be used safely and effectively at relapse.65,67,108 Immune modulatory drugs are commonly used in the treatment of relapsed/refractory patients with AL amyloidosis. Lenalidomide and pomalidomide grant hematologic response in approximately 50% of patients with generally low (∼15%) CR rates and can rescue patients who are refractory to alkylators, proteasome inhibitors, and other immunomodulatory drugs.109,110 Immunomodulatory drugs cause an increase in cardiac biomarkers that interferes with the assessment of cardiac response, and lenalidomide may worsen renal function in subjects with elevated proteinuria.111 The second-generation, orally available proteasome inhibitor ixazomib is active in patients previously exposed to bortezomib.92,112 Recent studies reported high CR/VGPR rates (∼80%) in relapsed/refractory patients with t(11;14) treated with venetoclax.27 Bendamustine can be considered in patients with IgM-AL amyloidosis. Dara, daratumumab; Dex, Dexamethasone; Ixa, Ixazomib; Len, lenalidomide; OS; overall survival; PFS, progression-free survival; Pom, pomalidomide.
Rescue treatment algorithm for AL amyloidosis. Relapsed/refractory patients should be enrolled in clinical trials whenever possible. Treating relapsed patients with a different class of agents than that used in upfront therapy is associated with prolonged PFS but has no impact on OS.107 Thus, if response to the previous line of therapy lasted at least 12 to 18 months, retreatment with the same drugs can be considered. Eligible patients who did not perform ASCT can be transplanted at relapse. At present, many relapsing patients have not been exposed to daratumumab, and this agent alone or combined with bortezomib or lenalidomide can be used safely and effectively at relapse.65,67,108 Immune modulatory drugs are commonly used in the treatment of relapsed/refractory patients with AL amyloidosis. Lenalidomide and pomalidomide grant hematologic response in approximately 50% of patients with generally low (∼15%) CR rates and can rescue patients who are refractory to alkylators, proteasome inhibitors, and other immunomodulatory drugs.109,110 Immunomodulatory drugs cause an increase in cardiac biomarkers that interferes with the assessment of cardiac response, and lenalidomide may worsen renal function in subjects with elevated proteinuria.111 The second-generation, orally available proteasome inhibitor ixazomib is active in patients previously exposed to bortezomib.92,112 Recent studies reported high CR/VGPR rates (∼80%) in relapsed/refractory patients with t(11;14) treated with venetoclax.27 Bendamustine can be considered in patients with IgM-AL amyloidosis. Dara, daratumumab; Dex, Dexamethasone; Ixa, Ixazomib; Len, lenalidomide; OS; overall survival; PFS, progression-free survival; Pom, pomalidomide.
The optimal goal of therapy is yet to be established and possibly varies during follow-up. In patients with this rapidly progressing disease, it is necessary to closely monitor (every 2 cycles) the outcome of therapy to promptly switch regimen and avoid further organ deterioration.61,81 A deep reduction of FLCs should be achieved very early,81 but ideal timing and depth vary based on disease severity and treatment regimen. With daratumumab, FLCs usually drop after the first administration.67 In patients with advanced cardiac involvement, an earlier (after 1 cycle) assessment of response is advisable to allow a rapid shift in nonresponders. This can be particularly relevant in subjects receiving daratumumab. At the end of frontline therapy, either CR or organ response should be reached, and the recently validated composite hematologic and organ response criteria can offer guidance in this assessment.82 However, in patients with an aggressive underlying clone (eg, with SLiM CRAB or high-risk myeloma cytogenetics) CR might be more obstinately pursued, as recently suggested by the Mayo Clinic group.69 In the longer term, biomarkers of organ involvement should progressively decrease and normalize, indicating amyloid FLC concentration is kept at a level that does not prevent recovery of organ dysfunction.50 In fit patients who fail to attain organ response despite CR, MRD assessment can be useful, and further therapy can be considered if MRD is detected.49 The role of MRD as an end point in AL amyloidosis should be explored in future studies.
Case 5: an older woman with lymphadenopathy and an IgM monoclonal protein
A 77-year-old woman was referred to a hematologist because of bilateral enlarged cervical lymph nodes. Subsequent investigations showed mediastinal and retroperitoneal lymphadenopathy (<2 cm). An IgMλ monoclonal component was detected. Blood tests were all normal. A lymph node biopsy showed amyloid deposits. No clonal lymphocytes or plasma cells were detected in her bone marrow. The patient was followed closely without therapy. After 2 years, bilateral, symmetrical, ascending, sensitive, peripheral neuropathy appeared, and the patient was referred to our center. The concentration of the monoclonal protein was 25 g/L, λ FLC level was 40 mg/L (dFLC, 24 mg/L; ratio, 0.40), hemoglobin level was 11.5 g/dL, and alkaline phosphatase level was 360 U/L (upper reference limit, 150 U/L) with normal bilirubin and aminotransferases. Testing for anti-myelin–associated glycoprotein and anti-ganglioside antibodies was negative. Nerve conduction studies showed a purely axonal pattern. There were no signs of amyloid cardiac or renal involvement. We repeated a bone marrow biopsy that showed a 7% clonal, CD20-positive lymphoplasmacytic infiltrate. The clone harbored both the MYD88L265P and the CXCR4 mutations. The patient was treated with bendamustine, rituximab, and dexamethasone, which was well tolerated, and after 4 cycles, a low dFLC response was reached (dFLC, 4 mg/L; ratio, 0.63; monoclonal component, 10 g/L). Two more cycles were performed without improvement of hematologic response while alkaline phosphatase normalized. The patient is hematologically and clinically stable 1 year after treatment discontinuation.
Comments on patient 5
IgM-related AL amyloidosis is a distinct clinical entity that accounts for 6% to 10% of all AL amyloidosis cases.83,84 Localized amyloid deposits are more common, as well as lymph nodes, peripheral nervous system, and lung involvement, and amyloid FLC levels tend to be lower. The level of cardiac biomarkers is prognostic, but liver involvement and peripheral neuropathy also predict a poor outcome and were incorporated in a staging system specific for IgM-AL amyloidosis.84 Peripheral neuropathy is frequently symptomatic, but it is not detected by nerve conduction studies. Having both peripheral nervous system and liver involvement, this patient classified stage III despite normal cardiac biomarkers.
In most patients with IgM-AL amyloidosis, the clone can be detected in the bone marrow at diagnosis. The underlying clone can be a pure PC (in ∼25% of cases) or a lymphoplasmacytic neoplasm (in ∼65% of cases) as in the present case, and this is relevant for the choice of treatment.85 Lymphoplasmacytic clones harbor the MYD88 (95%-97%), CXCR4 (30%-40%), ARID1A (17%), and CD79B (8%-15%) mutations. Patients with IgM-AL amyloidosis caused by pure plasma cell clones should be treated like patients with non–IgM-AL amyloidosis, whereas patients with lymphoplasmacytic clones are generally treated with rituximab-containing regimens, based on treatments developed in Waldenström macroglobulinemia. Rituximab can be combined with dexamethasone and cyclophosphamide, bortezomib, or bendamustine.86-88 Patients with IgM-AL amyloidosis can also successfully undergo autologous stem cell transplant.89 Ibrutinib can be an option in patients with favorable mutational pattern, but a first report highlighted a rather poor tolerability and scarce efficacy of this drug.90 Patients with IgM-AL amyloidosis have lower response rates and shorter survival than other patients with AL amyloidosis when adjusted to stage.85
Neuropathy was the main reason to start treatment in this subject. The differential diagnosis of monoclonal gammopathies of neurologic significance is challenging.91 In this patient, age was considered a contraindication to ASCT, and we preferred a bortezomib-free combination because of neuropathy. The validated response criteria of the International Society of Amyloidosis consistently predict survival and can be used in IgM-AL amyloidosis.84
Conclusion
After many years of empirical treatment, the therapy of patients with AL amyloidosis has now entered the realm of evidence-based medicine, with 3 international clinical trials completed in the last 2 years.44,73,92 We should now further exploit these achievements with new controlled trials based on validated end points. However, our evidence-based knowledge only covers a small part of the management of patients with AL amyloidosis. Daratumumab-bortezomib combinations can be used as frontline therapy in almost all patients, and the key eligibility criteria to safely proceed to ASCT are now shared by referral centers. However, important questions, such as best treatment at relapse, role of maintenance, and the most cost-effective approach in patients with specific chromosomal abnormalities still need to be answered in controlled studies. For these reasons, patients should still be referred to specialized centers and treated within clinical trials whenever possible. With a growing therapeutic armamentarium, survival has more than tripled; however, no progress was made in patients with stage IIIb. Amyloid-directed therapy is an appealing opportunity to accelerate recovery of organ function, particularly in these patients. New therapies are emerging, and more than 20 interventional trials are now recruiting. Immunotherapies showing promise in MM will hopefully be offered to patients with AL amyloidosis. The anti-CD38 antibody isatuximab showed encouraging results in a recent phase 2 clinical trial in relapsed/refractory patients (NCT03499808).93 Chimeric antigen receptor T cells, and bispecific T-cell engager antibodies and antibody–drug conjugates, such as belantamab mafodotin that is being evaluated in a European trial (NCT04617925), will allow targeting additional PC antigens. Finally, the continuous, stimulating progress in basic and translational research are offering new insights in the mechanisms of disease, and there is hope that new treatment targets will eventually be made available.
Acknowledgments
The authors received research support by a grant from CARIPLO “Harnessing the plasma cell secretory capacity against systemic light chain amyloidosis” (grant 2018-0257), a grant from the Italian Ministry of Health “Towards effective, patient-tailored anti-plasma cell therapies in AL amyloidosis: predicting drug response and overcoming drug resistance” (grant GR-2018-12368387), and by a grant from Cancer Research United Kingdom, Fundación Científica Asociación Española Contra el Cáncer, and Associazione Italiana per la Ricerca sul Cancro under the Accelerator Award 2017 Program “Early detection and intervention: understanding the mechanisms of transformation and hidden resistance of incurable haematological malignancies.”
Authorship
Contribution: G.P. and G.M. wrote the manuscript.
Conflict-of-interest disclosure: G.P. reports honoraria for lectures from and membership on advisory boards with Janssen and honoraria for lectures from Pfizer and Siemens. G.M. declares no competing financial interests.
Correspondence: Giampaolo Merlini, Amyloidosis Research and Treatment Center, Foundation IRCCS Policlinico San Matteo, Viale Golgi, 19, 27100 Pavia, Italy; e-mail: gmerlini@unipv.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal