

Key Points
Donor-derived mLSTs with native specificity for leukemia-expressed antigens can be expanded ex vivo.
Infusion of mLSTs after hematopoietic stem cell transplant is well tolerated and may contribute to prevention of relapse.

Abstract
Hematopoietic stem cell transplant (HSCT) is a curative option for patients with high-risk acute lymphoblastic leukemia (ALL), but relapse remains a major cause of treatment failure. To prevent disease relapse, we prepared and infused donor-derived multiple leukemia antigen–specific T cells (mLSTs) targeting PRAME, WT1, and survivin, which are leukemia-associated antigens frequently expressed in B- and T-ALL. Our goal was to maximize the graft-versus-leukemia effect while minimizing the risk of graft-versus-host disease (GVHD). We administered mLSTs (dose range, 0.5 × 107 to 2 × 107 cells per square meter) to 11 patients with ALL (8 pediatric, 3 adult), and observed no dose-limiting toxicity, acute GVHD or cytokine release syndrome. Six of 8 evaluable patients remained in long-term complete remission (median: 46.5 months; range, 9-51). In these individuals we detected an increased frequency of tumor-reactive T cells shortly after infusion, with activity against both targeted and nontargeted, known tumor-associated antigens, indicative of in vivo antigen spreading. By contrast, this in vivo amplification was absent in the 2 patients who experienced relapse. In summary, infusion of donor-derived mLSTs after allogeneic HSCT is feasible and safe and may contribute to disease control, as evidenced by in vivo tumor-directed T-cell expansion. Thus, this approach represents a promising strategy for preventing relapse in patients with ALL.
Introduction
Patients with acute lymphoblastic leukemia (ALL) who relapse after allogeneic hematopoietic stem cell transplant (HSCT) have a dismal prognosis, with few effective therapeutic options.1-3 Donor lymphocyte infusions (DLIs) may promote a graft-versus-leukemia (GVL) effect, but any potential benefit must be weighed against the risk of graft-versus-host disease (GVHD) caused by the alloreactive cells present in the infused product.4,5 If DLIs are to be used as an adjuvant that prevents rather than treats post-HSCT relapse, there is an urgent need to enhance their safety by minimizing alloreactive T cells and amplifying the tumor-specific T-cell content, thereby augmenting beneficial GVL activity.
To achieve this goal, we developed an ex vivo strategy to selectively enrich for donor-derived tumor-reactive populations. To maximize the therapeutic potential, we expanded T cells with reactivity for a cohort of leukemia-specific antigens that are frequently expressed in ALL - WT1, PRAME, and survivin.6-11 We reasoned that these multiple leukemia antigen–specific T cells (mLSTs) could be safely administered to all patients with high-risk B- or T-cell ALL after allogeneic HSCT, to prevent relapse. We now detail the outcomes of patients with B-ALL, T-ALL, or mixed phenotype acute leukemia (MPAL), who received mLSTs as an adjuvant treatment after HSCT.
Methods
Clinical study
The protocol was approved by the Baylor College of Medicine institutional review board; written informed consent was obtained from all participants or parents in accordance with institutional guidelines and the Declaration of Helsinki. Allo-HSCT recipients with B- or T-cell ALL or MPAL were eligible. Detailed inclusion and exclusion criteria are outlined in the protocol (registered at www.clinicaltrails.gov as #NCT02475707; supplemental Methods, avaible on the Blood Web site). Once enrolled, patients with engrafted transplants, at least 30 days past HSCT, received a single infusion of mLSTs at 1 of 3 dose levels (DLs; DL1, 0.5 × 107 cells per square meter; DL2, 1 × 107 per square meter; or DL3, 2 × 107 per square meter) and had the option to receive up to 6 additional infusions if they remained in complete remission (CR). None of the patients received lymphodepleting chemotherapy before the mLSTs.
Generation of mLSTs
mLSTs were generated as previously described.12 In brief, monocyte-derived dendritic cells were loaded with overlapping peptide libraries (pepmixes) spanning survivin, WT1, and PRAME and cocultured with donor peripheral blood mononuclear cells in T-cell medium supplemented with interleukin-7 (IL7), -12, -15, and -6. From day 10 and weekly thereafter, responder T cells were restimulated with pepmix-pulsed dendritic cells in the presence of IL15 or -2 until a sufficient number was achieved for patient infusion.
mLST characterization and immune monitoring
Details are found in supplemental Methods. In brief, mLSTs were phenotypically and functionally characterized using interferon γ (IFNγ) enzyme-linked immunospot (ELIspot), flow cytometry, and 51chromium-release assays.
Statistical analysis
Descriptive statistics were calculated, and clinical and correlative characteristics were summarized as the mean, standard deviation, standard error of the mean (SEM), median, and range. Dose escalation was guided by the modified continual reassessment method, as detailed in the “Study Design” of the clinical protocol in the supplemental Methods. Dose-limiting toxicity was defined as grade 3 or 4 GVHD or National Cancer Institute Common Toxicity Criteria grade 3, 4, or 5 toxicity that was infusion related and occurred within 4 weeks of infusion of mLSTs. The data cutoff date for analysis was 1 September 2021.
Results and discussion
Fifteen patients were enrolled in the study, and we successfully generated donor-derived mLSTs for each of these patients. Eleven patients (3 adults and 8 children) with B-ALL (n = 9), T-ALL (n = 1), or MPAL (n = 1) who were recipients of matched-sibling (n = 9), matched-family (n = 1), or haplo (n = 1) transplants, were infused with mLSTs (dose range, 0.5 × 107 to 2 × 107cells per square meter) at a median of 107 days after transplant (range, 48-167). The timing of the infusion was driven by mLST availability or the clinical status of the patient (eg, resolution of GVHD). Clinical characteristics and outcomes are detailed in Table 1. Four patients were not infused (supplemental Figure 1).
Characteristics and outcome of evaluable patients
| ID | Age/sex | Diagnosis; DS at time of mLST infusion | Prior treatments | Conditioning regimen; GVHD prophylaxis | HSCT donor | GVHD after HSCT, status before mLSTs | DL | Immune suppression at time of mLST | Day of mLSTs after HSCT | GVHD after mLST infusion | Post-mLST infusion relapse | Post-mLST infusion survival |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 41/M | Ph-like, poor risk cytogenetics; CR1* | Hyper-CVAD+ofatumumab ×5 cycles | TBI (12Gy)/CTX; tacro/MTX | MSD | None | 1 | Tacro | 66 | None | No | Alive at 47 mo |
| 3 | 17/F | Relapsed ALL; CR2, MRDneg | Completed therapy for HR- ALL per COG AALL1131→relapse→ reinduction per COG AALL1331HSCT | TBI (6Gy)/flu/ alemtuzumab; tacro | MSD | Acute GVHD grade 2; resolved | 1 | Tacro | 107 | None | Yes 6 mo | Died at 18 mo (relapse) |
| 4 | 14/M | Ph+ALL; CR2, MRDneg | Completed therapy for HR- ALL COG AALL1122 (dasatinib)→relapse → reinduction on COG AALL0434 (dasatinib)→HSCT | TBI (12Gy)/CTX; ara-C; tacro methylpred | MSD | Acute GVHD grade 2; resolved | 1 | Tacro | 127 | Chronic moderate GVHD 5 mo after infusion | No | Died at 9.5 mo (Varicella zoster infection) |
| 5 | 20/F | Ph+ALL; CR3, MRDneg | Induction and consolidation-COG AALL0622 (dasatinib)→MSDSCT→ relapse→ALLR3 reinduction chemotherapy→CD34+ top-off† | TBI (6Gy)/flu/ alemtuzumab; tacro | MSD | None | 1 | None‡ | 48 | None | No | Alive at 51 mo |
| 7 | 12/F | T-cell ALL; CR2, MRDneg | Completed therapy for T- ALL COG AALL0434→relapse→ reinduction on COG AALL1231 (bortezomib)→HSCT | TBI (12Gy)/CTX; tacro/MTX | MSD | None | 2 | None | 117 | None | No | Alive at 48 mo |
| 8 | 18/M | HR-ALL; CR1, MRDneg | Induction and consolidation COG AALL1131→primary induction failure→HSCT | TBI (12Gy)/CTX/ara-C; tacro/MTX | MSD | None | 2 | Tacro | 48 | None | No | Alive at 46 mo |
| 10 | 12/F | T-cell/ MPAL, FLT3+; CR1, MRDneg | Induction and consolidation chemotherapy COG AALL1231→HSCT as high risk | TBI (12Gy)/CTX; tacro/MTX | MSD | Acute GVHD Grade 2; resolved | 3 | Tacro, pred§ | 146 | None | No | Alive at 36 mo |
| 12 | 23/F | Ph+ALL, high-risk cytogenetics; CR3; MRDneg | Completed therapy for COG AALL08P1 (augmented BFM regimen)→relapse 8 y later→hyper-CVAD × 4 cycles→residual disease→CVAD+ inotuzumab mo 4 cycles+5 cycles of POMP→HSCT | TBI (2Gy) flu/mel; post-HSCT CTX tacro/MMF | Haplo | None | 3 | Tacro | 111 | None | Yes 10 mo | Alive at 24 mo |
| ID | Age/sex | Diagnosis; DS at time of mLST infusion | Prior treatments | Conditioning regimen; GVHD prophylaxis | HSCT donor | GVHD after HSCT, status before mLSTs | DL | Immune suppression at time of mLST | Day of mLSTs after HSCT | GVHD after mLST infusion | Post-mLST infusion relapse | Post-mLST infusion survival |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 41/M | Ph-like, poor risk cytogenetics; CR1* | Hyper-CVAD+ofatumumab ×5 cycles | TBI (12Gy)/CTX; tacro/MTX | MSD | None | 1 | Tacro | 66 | None | No | Alive at 47 mo |
| 3 | 17/F | Relapsed ALL; CR2, MRDneg | Completed therapy for HR- ALL per COG AALL1131→relapse→ reinduction per COG AALL1331HSCT | TBI (6Gy)/flu/ alemtuzumab; tacro | MSD | Acute GVHD grade 2; resolved | 1 | Tacro | 107 | None | Yes 6 mo | Died at 18 mo (relapse) |
| 4 | 14/M | Ph+ALL; CR2, MRDneg | Completed therapy for HR- ALL COG AALL1122 (dasatinib)→relapse → reinduction on COG AALL0434 (dasatinib)→HSCT | TBI (12Gy)/CTX; ara-C; tacro methylpred | MSD | Acute GVHD grade 2; resolved | 1 | Tacro | 127 | Chronic moderate GVHD 5 mo after infusion | No | Died at 9.5 mo (Varicella zoster infection) |
| 5 | 20/F | Ph+ALL; CR3, MRDneg | Induction and consolidation-COG AALL0622 (dasatinib)→MSDSCT→ relapse→ALLR3 reinduction chemotherapy→CD34+ top-off† | TBI (6Gy)/flu/ alemtuzumab; tacro | MSD | None | 1 | None‡ | 48 | None | No | Alive at 51 mo |
| 7 | 12/F | T-cell ALL; CR2, MRDneg | Completed therapy for T- ALL COG AALL0434→relapse→ reinduction on COG AALL1231 (bortezomib)→HSCT | TBI (12Gy)/CTX; tacro/MTX | MSD | None | 2 | None | 117 | None | No | Alive at 48 mo |
| 8 | 18/M | HR-ALL; CR1, MRDneg | Induction and consolidation COG AALL1131→primary induction failure→HSCT | TBI (12Gy)/CTX/ara-C; tacro/MTX | MSD | None | 2 | Tacro | 48 | None | No | Alive at 46 mo |
| 10 | 12/F | T-cell/ MPAL, FLT3+; CR1, MRDneg | Induction and consolidation chemotherapy COG AALL1231→HSCT as high risk | TBI (12Gy)/CTX; tacro/MTX | MSD | Acute GVHD Grade 2; resolved | 3 | Tacro, pred§ | 146 | None | No | Alive at 36 mo |
| 12 | 23/F | Ph+ALL, high-risk cytogenetics; CR3; MRDneg | Completed therapy for COG AALL08P1 (augmented BFM regimen)→relapse 8 y later→hyper-CVAD × 4 cycles→residual disease→CVAD+ inotuzumab mo 4 cycles+5 cycles of POMP→HSCT | TBI (2Gy) flu/mel; post-HSCT CTX tacro/MMF | Haplo | None | 3 | Tacro | 111 | None | Yes 10 mo | Alive at 24 mo |
ara-C, cytarabine; COG, Children’s Oncology Group; CTX, cytoxan; DL, dose level; DS, disease status; Flu, fludarabine; GVHD, graft-versus-host disease; Haplo, haploidentical donor; HSCT, hematopoietic stem cell transplant; hyper-CVAD, cyclophosphamide, vincristine, doxorubicin, and dexamethasone; Mel, melphalan; mLST, multiple leukemia-associated antigen specific T cells; MMF, mycophenolate mofetil; MPAL, mixed phenotype acute leukemia; MRDneg, minimal residual disease negative; MSD, matched sibling donor; MTX, methotrexate; Ph+ ALL, Philadelphia positive acute lymphoblastic leukemia; Ph-like, Philadelphia-like; POMP, 6-mercaptopurine+vincristine+methotrexate+prednisone; Pred, prednisone; Tacro, tacrolimus; TBI, total body irradiation.
MRD not available, normal cytogenetics.
Received mLST infusion following CD34+ stem cell top-off.
Received tyrosine kinase inhibitor (bosutinib, 3 wk post-mLST infusion for 1 y).
Dose: 0.1 mg/kg.
The 15 mLST lines generated for clinical use were composed of CD3+ T cells (mean ± SEM: 95.1% ± 1.9%), with a mixture of CD4+ (22.8% ± 6.3%) and CD8+ (52.5% ± 5.3%) cells, which expressed central (CD45RO+/CD62L+; 13.5% ± 2.8%) and effector memory markers (CD45RO+/CD62L−; 56.4% ± 3.8%) (Figure 1A). mLST lines recognized the targeted antigens PRAME (range, 0-1554; mean ± SEM: 243 ± 103 spot-forming cells (SFCs)/2 × 105), WT1 (range, 0-2088; 227 ± 134 SFCs/2 × 105), and survivin (0-394; 57 ± 31 SFCs/2 × 105) as judged by IFNγ ELIspot assays (Figure 1B). No alloreactivity was observed; none of the lines reacted against nonmalignant patient-derived cells (3.6% ± 0.8% specific lysis; effector/target ratio, 20:1), a product release criterion (Figure 1C).
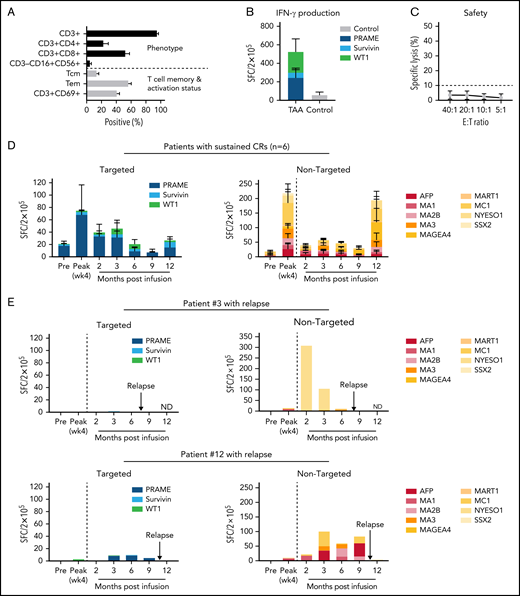
Characterization and in vivo fate of donor-derived mLSTs. (A-C) Characterization of donor-derived mLSTs. (A) Phenotype and memory/activation profile. (B) Specificity of mLSTs as measured by IFNγ ELIspot for 15 products generated using all 3 antigens as a stimulus. Data are shown as mean SFCs ± SEM/2 × 105 and each color represents an individual antigenic specificity. (C) Lack of in vitro mLST cytolytic activity against normal recipient cells at effector/target ratios from 40:1 to 5:1. (D) In vivo behavior of mLSTs in patients who remain in remission (n = 6). Expansion of T cells specific for targeted TAAs (left) and other nontargeted TAAs (right) in patients who responded to therapy. Results are reported as mean SFCs ± SEM/2 × 105 at each specified time point. (E) In vivo behavior of mLSTs in patients who relapsed. Lack of T-cell expansion against either targeted (left) or nontargeted TAAs (right) immediately after infusion in patients 3 and 12 who eventually relapsed. Results are reported as SFC/2 × 105 at each specified time point. Arrows indicate the time of relapse.
Characterization and in vivo fate of donor-derived mLSTs. (A-C) Characterization of donor-derived mLSTs. (A) Phenotype and memory/activation profile. (B) Specificity of mLSTs as measured by IFNγ ELIspot for 15 products generated using all 3 antigens as a stimulus. Data are shown as mean SFCs ± SEM/2 × 105 and each color represents an individual antigenic specificity. (C) Lack of in vitro mLST cytolytic activity against normal recipient cells at effector/target ratios from 40:1 to 5:1. (D) In vivo behavior of mLSTs in patients who remain in remission (n = 6). Expansion of T cells specific for targeted TAAs (left) and other nontargeted TAAs (right) in patients who responded to therapy. Results are reported as mean SFCs ± SEM/2 × 105 at each specified time point. (E) In vivo behavior of mLSTs in patients who relapsed. Lack of T-cell expansion against either targeted (left) or nontargeted TAAs (right) immediately after infusion in patients 3 and 12 who eventually relapsed. Results are reported as SFC/2 × 105 at each specified time point. Arrows indicate the time of relapse.
Ten patients received a single infusion of mLSTs (treating physicians or families opted not to administer additional infusions), whereas 1 patient (who developed mixed chimerism) received a total of 4 infusions to prevent overt relapse. All infusions were safe and well tolerated. No patient developed acute post-mLST GVHD. A single patient, with a history of acute GVHD early after transplant that had resolved with therapy before mLSTs, developed moderate chronic GVHD 5 months after infusion. There were no instances of cytokine release syndrome, neurotoxicity, or grade ≥3 adverse events that were attributable to mLSTs.
Three of the 11 patients infused were not evaluable, as each received >0.5 mg/kg prednisone or equivalent within 4 weeks of infusion (supplemental Table 1). Two received stress doses of hydrocortisone for the treatment of septic shock, and 1 received prednisone for elevated liver transaminases attributable to acetaminophen overdose (>4 g/d). Liver enzymes normalized once acetaminophen was discontinued.
The remaining 8 infused patients who received mLSTs as adjuvant treatment while in CR after HSCT were evaluable for long-term safety and efficacy. Six remained in minimal residual disease–negative CR at a median of 46.5 months after infusion (range, 9.5-51 months). Two patients relapsed; 1 of those (patient 3) received a reduced-intensity conditioning regimen due to underlying comorbidities before transplant and was infused with mLSTs 3 months after HSCT. She developed mixed chimerism 6 weeks later, subsequently received 3 additional infusions at monthly intervals, and remained in CR for 6 months from the initial mLST infusion date but ultimately relapsed and died. The second patient (patient 12) underwent a reduced-intensity conditioning haploidentical HSCT and received mLSTs 4 months after HSCT but relapsed 10 months later. Subsequent treatment with tisagenlecleucel was unsuccessful but with salvage chemotherapy followed by a second allogeneic HSCT, she again achieved CR.
To investigate the contribution of the infused mLSTs in sustaining remission we evaluated the frequency of tumor-reactive T cells in peripheral blood before and after infusion. We analyzed both T-cell responses to the antigens targeted by the infused mLSTs (WT1, survivin, PRAME), as well as against a range of other nontargeted tumor-associated antigens (TAAs) including SSX2; MAGE-A4 -A1, -A2B, and -C1; MART1; AFP; and NYESO1. We reasoned that detection of such cells could be indicative of an active GVL effect mediated by mLSTs, producing in vivo TAA spreading, thereby enhancing the antitumor benefits of our therapy. All patients, who achieved a long-term CR, showed an expansion of tumor-specific cells (both infused and endogenous), which peaked within 4 weeks of infusion (Figure 1D; supplemental Figure 2). By contrast, in the 2 patients who relapsed, we saw no evidence of this immune signature (Figure 1E). Supplemental Figure 3 details the results of the nonevaluable patients.
In summary, the preparation and infusion of donor-derived mLSTs in patients with ALL after allogeneic HSCT is feasible and safe, and, as evidenced by in vivo tumor-directed T-cell expansion and antigen spreading in patients, may contribute to disease control. Early relapses after HSCT are associated with poor outcome, and their prevention may improve survival after transplant.13,14 This strategy represents a promising addition to approaches for prophylaxis for relapse after HSCT and warrants larger, controlled studies.
Acknowledgments
The authors thank patients and their families who participated in this trial; the clinicians, staff, and integral personnel in the clinic and the laboratory; and, specifically, Deborah Lyon for quality assurance and quality control and Walter Mejia for assisting with formatting the figures.
This work was supported by the Leukemia and Lymphoma Society Specialized Center of Research (SCOR) awards 7001-14 (H.E.H.), a Cookies for Cancer Research grant (A.M.L.), Cancer Prevention and Research Institute of Texas (CPRIT) Texas Access to Cancer Cell Therapies (TACCT) grant RP180785, a CPRIT Early Career Clinical Investigator Award RP200584 (P.D.L.), an Edward P. Evans Foundation Discovery Research Grant (P.D.L.) 2018, and the Dan L. Duncan Comprehensive Cancer Center for application of the shared resources from a support grant from the National Institutes of Health (NIH), National Cancer Institute (P30CA125123). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: A.M.L., S.G., P.D.L., and S.N. conceived and designed the study; B.G., C.R., and N.L. provided administrative, regulatory, and quality assurance support; S.N., P.D.L., R.T.K., S.G., G.C., L.H., R.A.K., C.M., P.T., and B.O. recruited and cared for the patients; W.T. and M.W. performed the statistical analysis; S.V., I.T., M.K., and A.W. generated mLSTs and performed correlative studies; S.N., P.D.L., A.M.L., H.E.H., M.K.B., S.V., C.M.R., and J.F.V. analyzed and provided input on data interpretation; S.N., P.D.L., A.M.L., and S.V. wrote the manuscript; and all authors approved the final draft of the manuscript and are accountable for all aspects of the work.
Conflict-of-interest disclosure: Marker Therapeutics has licensed this technology from Baylor College of Medicine. B.G. owns QBRegulatory Consulting which provides consulting services to AlloVir, Tessa, LOKON, and Marker. A.M.L., J.F.V., M.K.B., C.M.R., and H.E.H. are cofounders and equity holders in AlloVir Inc and Marker Therapeutics. A.M.L., S.V., M.K., and A.W. are consultants for AlloVir Inc. J.F.V. is an employee of Marker Therapeutics, which aspires to commercialize the described approach. M.K.B. is the founder member of and serves on the advisory board for Tessa Therapeutics. He is on the advisory boards of Bluebird Bio and Turnstone and the science advisory boards of Memgen, KUUR, Bellicum Pharmaceuticals, Tscan, Poseida, Abintus, Allogene, and Walking Fish and has stock option in Allogene and Walking Fish. C.M.R. is the spouse of M.K.B and declares the same conflicts as M.K.B. She is also on the science advisory board of and receives funding support from Tessa Therapeutics. H.E.H. has served on advisory boards for Gilead Biosciences, Novartis, PACT Pharma, Mesoblast, Kiadis, GSK, Fresh Wind Biotherapies, Takeda, and Tessa Therapeutics and has received research support from Tessa Therapeutics and Kuur Therapeutics. L.H. is on the advisory board of Incyte for GVHD. P.D.L. served on an advisory board for Karyopharm Therapeutics. N.L. is consultant to Tessa Therapeutics. S.G. has a consulting agreement with Tessa Therapeutics, is a compensated member of the data and safety monitoring board of Immatics, has received honoraria from Tidal, Catamaran Bio, and Novartis within the past 2 years, and has received past research support from Tessa Therapeutics. S.N., I.T., C.R., W.T., M.W., G.C., R.T.K., R.A.K., C.M., P.T., and B.O. report no competing financial interests.
The current affiliation for S.N. and S.G. is Department of Bone Marrow Transplant and Cellular Therapy, St Jude Children’s Research Hospital, Memphis, TN.
The current affiliation for P.T. is Division of Pediatrics, The University of Texas MD Anderson Cancer Center, Houston, TX.
Correspondence: Swati Naik, Department of Bone Marrow Transplantation and Cellular Therapy, St Jude Children’s Research Hospital, 262 Danny Thomas Pl, MS 1130, Memphis, TN 38105; e-mail: swati.naik@stjude.org; and Premal D. Lulla, Center for Cell and Gene Therapy, Feigin Center FC1780.06, 1102 Bates Ave, Houston, TX 77030; e-mail: lulla@bcm.edu.
For data sharing, please contact the corresponding authors, Swati Naik and/or Premal Lulla.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal