

Key Points
Basal protein phosphorylation is higher in UM-CLL.
MARCKS regulates central signaling pathways and affects response to acalabrutinib.

Abstract
Bruton tyrosine kinase (BTK) inhibitors are highly active drugs for the treatment of chronic lymphocytic leukemia (CLL). To understand the response to BTK inhibitors on a molecular level, we performed (phospho)proteomic analyses under ibrutinib treatment. We identified 3466 proteins and 9184 phosphopeptides (representing 2854 proteins) in CLL cells exhibiting a physiological ratio of phosphorylated serines (pS), threonines (pT), and tyrosines (pY) (pS:pT:pY). Expression of 83 proteins differed between unmutated immunoglobulin heavy-chain variable region (IGHV) CLL (UM-CLL) and mutated IGHV CLL (M-CLL). Strikingly, UM-CLL cells showed higher basal phosphorylation levels than M-CLL samples. Effects of ibrutinib on protein phosphorylation levels were stronger in UM-CLL, especially on phosphorylated tyrosines. The differentially regulated phosphopeptides and proteins clustered in pathways regulating cell migration, motility, cytoskeleton composition, and survival. One protein, myristoylated alanine-rich C-kinase substrate (MARCKS), showed striking differences in expression and phosphorylation level in UM-CLL vs M-CLL. MARCKS sequesters phosphatidylinositol-4,5-bisphosphate, thereby affecting central signaling pathways and clustering of the B-cell receptor (BCR). Genetically induced loss of MARCKS significantly increased AKT signaling and migratory capacity. CD40L stimulation increased expression of MARCKS. BCR stimulation induced phosphorylation of MARCKS, which was reduced by BTK inhibitors. In line with our in vitro findings, low MARCKS expression is associated with significantly higher treatment-induced leukocytosis and more pronounced decrease of nodal disease in patients with CLL treated with acalabrutinib.
Introduction
Therapy of chronic lymphocytic leukemia (CLL) has changed over the years. As we gain more knowledge of CLL pathophysiology, new therapies targeting central pathways in malignant cells have entered the clinic.1,2 The course of CLL is heterogeneous and depends on a variety of risk factors, among them genetic aberrations and immunoglobulin heavy-chain variable region (IGHV) mutational status.3-5 Mutational status is determined based on identity to the germline IGHV sequence with 98% being used as the cutoff.6 Patients with unmutated IGHV CLL (UM-CLL) often exhibit a more aggressive disease course than patients with mutated IGHV CLL (M-CLL).3 The B-cell receptor (BCR) of UM-CLL cells is polyreactive and the level of BCR signaling is increased, whereas antigen binding to the BCR and its subsequent activation is more limited in M-CLL cells.7-9 Antigen-dependent and -independent BCR signaling promotes cell survival, proliferation, and migration via a complex network of signaling cascades.10
Recently, Xu et al identified the myristoylated alanine-rich C-kinase substrate (MARCKS) as a regulator of BCR lateral mobility and clustering.11 MARCKS, a protein kinase C (PKC) substrate, resides at the membrane, sequestering phosphatidylinositol-4,5-bisphosphate (PIP2).12-14 PIP2 is relevant for central signaling pathways like phosphatidylinositol 3-kinase (PI3K) signaling.15 MARCKS is involved in regulation of tumor invasiveness and has predominantly been associated with solid tumors.16-18
Bruton tyrosine kinase (BTK) is a central molecule in the BCR-signaling cascade and has proven to be an efficient therapeutic target. Ibrutinib was the first BTK inhibitor available and is licensed for both frontline therapy and relapsed/refractory CLL.19 Recently, second-generation BTK inhibitors like acalabrutinib have been developed that are highly selective for BTK, reducing off-target effects.20
To decipher the underlying mechanism, we performed comparative (phospho)proteomic analyses of UM-CLL and M-CLL cells. Several prior reports that focused on selected proteins identified differential pathways in UM-CLL, leading to a survival advantage of those cells.21,22 In some reports, the total proteome has been investigated.23-28 Johnston et al analyzed differences of the proteome in CLL cells and healthy B cells.27 Eagle et al reported distinct expression of proteins involved in migration and motility derived from analysis by isobaric tags for relative and absolute quantification (iTRAQ).28 Perrot et al found distinct expression of proteins involved in signal transduction, immune response regulation, and cell growth. They focused on changes in protein expression after BCR stimulation in 3 patients with M-CLL and 3 patients with UM-CLL, and found differential expression of proteins involved in BCR signaling like LSP1 and HNRPK.29
In contrast to proteomic analyses, little is known about the phosphoproteome (p-proteome). For example, O’Hayre et al performed phosphoproteomic analyses on high-risk CLL samples after stimulation with CXCL12 in 1 patient and compared these findings with 4 other patients, but did not analyze differences based on IGHV.30
Here, we used stable isotope labeling and iTRAQ and titanium dioxide (TiO2)-based phosphopeptide enrichment to obtain a comprehensive picture of the proteome and p-proteome of 3 UM-CLL and 3 M-CLL samples.
Material and methods
Patient samples
After informed consent according to the Helsinki protocol was given, blood was obtained from patients with CLL. CLL cells were negatively purified using Rosette Sep Human B-cell enrichment Cocktail (StemCell Technologies, Vancouver, BC, Canada) as described before.31-34 This study was approved by the Ethics Committee of the University of Cologne.
Quantitative proteome and p-proteome analysis
For phosphoproteomic analysis, we chose 4 different conditions: (1) untreated, (2) ibrutinib treatment, (3) stimulation with anti-immunoglobulin M (anti-IgM) beads, and (4) ibrutinib treatment plus stimulation with anti-IgM beads. A total of 1 × 108 isolated CLL cells per condition were used. Ibrutinib treatment (1 µM; Selleckchem, Houston, TX) was performed for 3 minutes, followed by stimulation with anti-IgM beads (100 µg/mL; Irvine Scientific, Santa Ana, CA) for 20 minutes. In line with our previous publications,31,33,35,36 we used IgM beads (rabbit anti-human IgM Immunobeads; Irvine Scientific). Samples for proteomic analysis were derived from the untreated cells. Instantly after treatment, cells were lysed in radioimmunoprecipitation assay buffer (see “Western blotting and immunodetection”) and frozen by liquid nitrogen.
Per sample, 150 µg of total protein was digested using trypsin and a modified filter-assisted sample preparation protocol.37,38 Digests were controlled using a monolithic high-performance liquid chromatography setup39 and samples were iTRAQ 8-plex labeled, yielding iTRAQ sets 1 to 3 (see Figure 1).
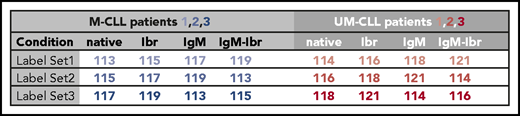
Prior to multiplexing the iTRAQ sets, 5 µg of labeled peptide of all untreated samples (mutated plus unmutated) was pooled to compose iTRAQ set 4 (see Figure 2). This set of untreated samples was used for proteomic analysis.

iTRAQ set 4 was fractionated by high pH reversed-phase chromatography and used for global proteome quantification, whereas iTRAQ sets 1 to 3 were each multiplexed and individually subjected to TiO2-based phosphopeptide enrichment and hydrophilic interaction liquid chromatography (HILIC) fractionation.40,41
All fractions were analyzed by nanoscale liquid chromatography coupled to tandem mass spectrometry (MS/MS) on Q Exactive or Q Exactive Plus Mass Spectrometers (both Thermo Fisher Scientific, Waltham, MA) in data-dependent acquisition mode. Elevated charge states of iTRAQ-labeled peptides were compensated for using 10% (v/v) NH4OH, as described previously.42
All data were analyzed using Proteome Discoverer (Thermo Fisher Scientific) and the false discovery rate was set to 0.01. Only phosphorylation sites with phosphoRS probabilities >90%, and only proteins quantified with at least 2 unique peptides were considered for quantitation. Data were normalized and scaled as previously described,41 and ratios were calculated using the 3 biological replicates. To compensate for intragroup heterogeneity, P values were determined using the moderated Student t test as proposed by Kammers et al and the Limma R package.43-45 Proteins being >2 σ away from the median ratio and having a value of P < .05 were considered as up/downregulated (supplemental Table 1, available on the Blood Web site). For the more dynamic p-proteome data, phosphopeptides being >3 σ away from the median ratio and having a P < .05 were considered as regulated (supplemental Tables 2 and 3).46
Further details are given in supplemental Methods.
MARCKS expression in B-cell subsets and CLL cells
In short, RNA was extracted from 10 000 cells by the Gentra Purescript protocol (Gentra) and processed by MessageAmp II aRNA Amplification and MessageAmp II Biotin Enhanced Kits (Ambion); 50 ng of RNA was processed with the OVATION Pico WTA System, the WT Ovation Exon Module, and the Encore Biotin Module (NuGen), and hybridized to HG U133 2.0 Plus Arrays. Data was normalized by variance-stabilizing normalization and corrected for batch effect by ComBat software.47
RNA-seq and analysis in acalabrutinib-treated patients with CLL
RNA-sequencing (RNA-seq) data from acalabrutinib-treated patients were previously published.48 Response parameters were compared between patient subgroups with below median and equal to or above median MARCKS expression at baseline.
RNA isolation for MARCKS reverse transcription polymerase chain reaction (PCR)
Total RNA isolation from cell pellets was performed by using TRIzol (Invitrogen, Darmstadt, Germany) according to the manufacturer’s instructions. All of the preparation and handling steps of RNA took place in a laminar flow hood, under RNA-free conditions. The isolated RNA was dissolved in 21 µL of RNAse-/DNAse-free water and stored at −80°C until used. RNA concentrations and ratios were determined photometrically (NanoDrop 1000; Peqlab, Erlangen, Germany).
Reverse transcription
Of the isolated total RNA, 1 µg was used for complementary DNA (cDNA) generation by the Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannheim, Germany). Reactions were set up and run according to the manufacturer’s instructions; thereafter, samples were kept at −20°C until use in quantitative PCR (qPCR).
MARCKS qPCR
qPCR was performed using the 7900HT Fast Real-Time PCR System (Thermo Fisher Scientific). The primers and the hybridization probes used were designed by ProbeFinder (Roche Diagnostics, Rotkreuz, Switzerland) and synthesized by Sigma-Aldrich (St. Louis, MO). qPCR was performed in a total volume of 20 µL. For the PCR, 5 µL of cDNA was placed into a 15-µL reaction volume containing 0.2 µL of the sense primer (1 µM), 0.2 µL of the antisense primer (1 µM), 4 µL of the TaqMan Fast Advanced Master Mix (2× concentration; Thermo Fisher Scientific), and 0.2 µL of the probe (1 µM); double-distilled water was added to the final volume. PCR was initiated with a 10-minute denaturation step at 95°C and terminated with a 30-second cooling step at 40°C. The cycling protocol consisted of a denaturation step at 95°C for 15 seconds and annealing at 60°C for 1 minute, repeated 40 times. Fluorescence detection was performed at the end of each annealing step for 1 second.
For quantification, an external calibration curve was obtained by using external standard cDNAs. The following Universal Probe Libraries (Roche Diagnostics, Rotkreuz, Switzerland) were used: probe #7 (reference [REF] 4685059001) for MARCKS, probe #11 (REF 4685105001) for ubiquitin C (UBC), and probe #73 (REF 4688961001) for HPRT1. The primer sequences for real-time PCR were as follows: MARCKS-specific primers (5′-ATGGGTGCCCAGTTCTCC-3′ [forward] and 5′-TTTACCTTCACGTGGCCATT-3′ [reverse]), HPRT1-specific primers (5′-TGACCTTGATTTATTTTGCATACC-3′ [forward] and 5′-CGAGCAAGACGTTCAGTCCT-3′ [reverse]) and UBC-specific primers (5′-GGAAGGCATTCCTCCTGAT-3′ [forward] and 5′-CCCACCTCTGAGACGGAGTA-3′ [reverse]) amplified fragments of the full-length transcripts. Results were normalized to HPRT1 and UBC. Log-fold change was calculated as described by Schmittgen et al.49
Western blotting and immunodetection
Treatments
To verify the experimental setup for phosphoproteomic analysis by immunoblot, 3 × 107 CLL cells were pretreated with ibrutinib (1 µM) or idelalisib (1 µM) or dimethyl sulfoxide for 3 minutes and subsequently stimulated with anti-IgM beads or left untreated. Samples were subjected to immunoblotting to analyze phosphorylation of BCR-related proteins in response to these stimuli.
To investigate phosphorylation of Akt after CXCL12 (Bio-Techne, Minneapolis, MN) treatment, 3 × 106 OSU-CLL cells were treated with CXCL12 (100 ng/mL) for 2 minutes after 3 hours of serum starvation.
CLL cells were seeded on CD40L feeder cells or treated with anti-IgM beads (100 µg/mL) for 24 hours to investigate MARCKS expression in response to these stimuli.
A total of 3 × 107 CLL cells, preincubated for 24 hours on CD40L feeder cells, were treated with anti-IgM beads (100 µg/mL) for 30 minutes after pretreatment with 1 µM ibrutinib for 1 hour to analyze phosphorylation of MARCKS.
CD40L+ NIH 3T3 mouse fibroblasts, irradiated and preincubated for 24 hours, were used for CD40L stimulation of CLL cells.
A total of 3 × 106 OSU-CLL cells were treated with 1 µM ibrutinib, 1 µM acalabrutinib, or 1 µM idelalisib for 1 hour to analyze MARCKS phosphorylation.
Immunoblot
Cells were lysed in radioimmunoprecipitation assay buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]; pH 8) with complete miniprotease inhibitor (Sigma-Aldrich, St Louis, MO), mixed with SDS-loading buffer (60 mM Tris-HCl, pH 6.8, 3.3% SDS, 20 mM dithiothreitol, 0.01% bromophenol blue), and then heated to 95°C for 5 minutes. Next, samples were subjected to gel electrophoresis at 180 V for 2 hours and then transferred to a Protean nitrocellulose membrane (0.45-µM pore size; Whatman, Little Chalfont, United Kingdom) by tank-blotting for 1 hour at 80 V. Blocking of membranes was performed for 1 hour at room temperature in 5% milk/Tris-buffered saline (TBS). The respective primary antibodies for MARCKS (clone D88D11; Cell Signaling, Danvers, MA/Merck Millipore, Burlington, MA), phosphorylated (p) MARCKS (pMARCKS) S167/170 (clone D13E4), pAkt S473, Akt (clone C67E7), phosphorylated extracellular signal-regulated kinase 1/2 (pERK 1/2; T202/Y204), pBTK Y223, phosphorylated phospholipase Cγ2 (pPLCγ2) Y1217, and pSyk Y525/526 (all Cell Signaling) were diluted in 5% milk/TBS (MARCKS [Cell Signaling], 1/500; pAkt, 1/1000) or 5% bovine serum albumin/TBS (MARCKS [Merck Millipore], 1/1000; pMARCKS, 1/1000; Akt, 1/2000; pBTK Y223, 1/1000; pERK, 1/1000; pPLCγ2, 1/1000; pSyk, 1/1000) and incubated overnight at 4°C. For detection of primary antibodies, specific IRDye secondary antibodies were used (LI-COR, Lincoln, NE). An antibody against glyceraldehyde-3-phosphate dehydrogenase (14C10; Cell Signaling) served as housekeeping gene-expression control. Western blot detection and density measurements were performed on the Odyssey infrared imaging system (LI-COR).
Transfections and CRISPR/Cas9
For MARCKS knockdown, 5 × 106 OSU-CLL cells were nucleofected with 100 pM specific ON-TARGETplus SMARTpool small-interfering RNA (siRNA; Dharmacon, Lafayette, CO). Nontargeting ON-TARGETplus siCONTROL siRNA (Dharmacon) was used as a negative control. The nucleofection was performed using program X01 at the Amaxa Nucleofector (Lonza, Basel, Switzerland). MARCKS knockout (KO) OSU-CLL cells were generated as described before.50 The short DNA sequences needed to perform clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) were designed using the Massachusetts Institute of Technology (MIT) online design tool (MARCKS_Ex_1_1 CACCGCCCGTCGTTACACCAACCCG).
Migration assay
Migration assays were performed in Transwell plates (8 µm; Corning, New York, NY) using RPMI 1640 medium (Thermo Fisher Scientific) without additives. CXCL12 (100 ng/mL) was added to the lower chamber. A total of 2 × 105 serum-starved OSU-CLL cells were placed in the upper wells. After 4 hours, migrated OSU-CLL cells were counted using MACSQuant VYB (Miltenyi Biotec, Bergisch Gladbach, Germany).
Results
Global proteomics in M-CLL vs UM-CLL reveal differences in cell death, growth, and cell motility
Our objective was to perform a quantitative p-proteome analysis of primary CLL samples and to investigate the differences between M-CLL and UM-CLL. Therefore, we aimed to elucidate the impact of ibrutinib treatment and/or BCR stimulation on the p-proteome in both subgroups. To establish our experimental setup, we first investigated the proteome and p-proteome of 4 pooled normal B-cell samples and 4 pooled M-CLL and UM-CLL samples, respectively. Indeed, we found proteins that are typically overexpressed in CLL cells, like ζ-chain–associated protein kinase 70 (ZAP70), FcµR, CD5, or LYN (data not shown) in the analyzed CLL samples. Furthermore, we defined an incubation time of 20 minutes for BCR stimulation to be suitable for quantitative phosphoproteomics based on initial experiments that showed no significant impact on protein expression (data not shown) but a strong effect on BCR signaling (supplemental Figure 1A-B). After establishing the experimental setup (Figure 3A), we analyzed the proteome of UM-CLL (n = 3) cells in comparison with M-CLL (n = 3) cells. Proteins that were >2 σ away from the median and having a P < .05, were considered as up/downregulated (supplemental Table 1). Of 3466 quantified proteins (Figure 3B), 83 (2.8%) showed a differential expression between the 2 subgroups (P ≤ .05; log2 fold change, 0.4). In UM-CLL, 45 of 83 proteins were upregulated and 38 proteins were downregulated in comparison with M-CLL (Figure 3C; supplemental Table 1). Ingenuity pathway analysis was used to assign the identified proteins to cellular proteins, such as migration and motility or survival and cell death (Figure 3D-F; supplemental Figure 2). The clinical significance of some of the proteins we identified, for example, ZAP70 or T-cell leukemia 1 (TCL1), has been reported before (Figure 3D,F).51-53
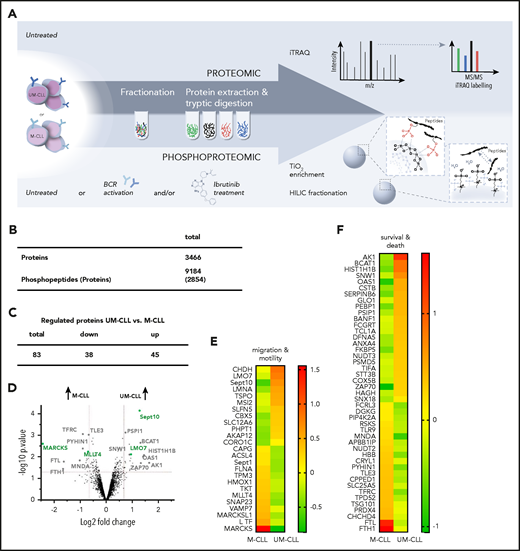
Analysis of proteomes and p-proteomes in M-CLL and UM-CLL cells. (A) Experimental setup for the mass spectrometric analysis of proteins and phosphoproteins from primary CLL samples involving iTRAQ labeling (proteome) and TiO2 enrichment plus HILIC fractionation (p-proteome). Phosphoproteomic analysis was performed under 4 different conditions: (1) native, (2) stimulation with anti-IgM beads, (3) ibrutinib (Ibr) treatment, and (4) ibrutinib treatment plus stimulation with anti-IgM beads. Proteome analysis was performed in untreated cells. (B) Number and regulation of isolated proteins and phosphopeptides. (C) Number of proteins with distinct expression in untreated UM-CLL (vs M-CLL). (D) Volcano plot of protein expression in untreated UM-CLL vs M-CLL cells (gray, proteins associated with cell death and survival; green, proteins associated with cell migration and motility). Negative log2 fold change: lower expression in UM-CLL, higher expression in M-CLL; positive log2 fold change: higher expression in UM-CLL, lower expression in M-CLL. (E-F) Heat maps representing expression levels of the most relevant regulated proteins in untreated UM-CLL vs M-CLL. The ratio of UM-CLL to M-CLL was calculated using the 3 biological replicates per group.
Analysis of proteomes and p-proteomes in M-CLL and UM-CLL cells. (A) Experimental setup for the mass spectrometric analysis of proteins and phosphoproteins from primary CLL samples involving iTRAQ labeling (proteome) and TiO2 enrichment plus HILIC fractionation (p-proteome). Phosphoproteomic analysis was performed under 4 different conditions: (1) native, (2) stimulation with anti-IgM beads, (3) ibrutinib (Ibr) treatment, and (4) ibrutinib treatment plus stimulation with anti-IgM beads. Proteome analysis was performed in untreated cells. (B) Number and regulation of isolated proteins and phosphopeptides. (C) Number of proteins with distinct expression in untreated UM-CLL (vs M-CLL). (D) Volcano plot of protein expression in untreated UM-CLL vs M-CLL cells (gray, proteins associated with cell death and survival; green, proteins associated with cell migration and motility). Negative log2 fold change: lower expression in UM-CLL, higher expression in M-CLL; positive log2 fold change: higher expression in UM-CLL, lower expression in M-CLL. (E-F) Heat maps representing expression levels of the most relevant regulated proteins in untreated UM-CLL vs M-CLL. The ratio of UM-CLL to M-CLL was calculated using the 3 biological replicates per group.
Our analysis identified novel differentially expressed proteins in M-CLL and UM-CLL. Proteins involved in cellular metabolism such as cytochrome c oxidase subunit 5B (COX5B) and adenylate kinase 1 (AK1) were especially upregulated in UM-CLL cells (Figure 3D,F). For COX5B it is known that overexpression is associated with various malignancies and poor prognosis.54-57 Downstream targets of the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2), like heme oxygenase 1 (HMOX1), ferritin light chain (FTL), and ferritin heavy chain 1 (FTH1) showed distinct expression profiles. These proteins are involved in protecting cells from oxidative stress,58 and their expression was higher in M-CLL (Figure 3D-F).
In general, our analyses revealed that basal expression of proteins associated with cell death, growth and survival, and proteins involved in cell motility and migration were different between M-CLL and UM-CLL, in line with data from other analyses for proteome analysis.28 For example, expression of MARCKS showed a striking difference between UM-CLL (low expression) and M-CLL (high expression) (Figure 3D-E; supplemental Figure 2). In malignant cells, MARCKS is involved in regulation of migration and motility/invasiveness, but the functional role of MARCKS has not been investigated in CLL so far.16,17,59,60
Phosphoproteomic analysis reveals higher basal phosphorylation in UM-CLL and clusters in pathways associated with migration and motility
Our next aim was to analyze and compare the p-proteome of M-CLL and UM-CLL. To validate our treatment conditions, we proved efficacy and optimal timing of BCR stimulation and BTK inhibition by immunoblot (supplemental Figure 1). In line with previously published data from Chen et al,9 phosphorylation of Syk and PLCγ was higher in unmutated CLL cells (supplemental Figure 1). In the phosphoproteomic analysis, we identified 9184 phosphopeptides in M-CLL and UM-CLL cells, which correspond to 2854 individual phosphoproteins (Figure 3B; supplemental Tables 2 and 3). Only peptides detected in 2 of 3 patients were included. Strikingly, UM-CLL cells exhibited increased basal phosphorylation compared with M-CLL cells (Figure 4B). One hundred eight phosphopeptides (92 proteins) of 129 differentially regulated phosphopeptides (109 proteins) (P ≤ .05; log2 fold change, 1) were phosphorylated more in UM-CLL, whereas total protein levels were similar in both subgroups (Figure 4A; supplemental Table 2).
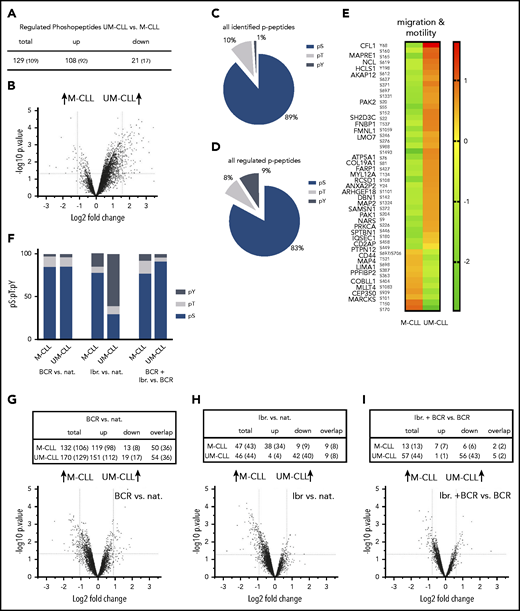
p-proteome analyses reveal distinct phosphorylation patterns in UM-CLL and M-CLL. (A) Number of differentially regulated p-peptides in untreated UM-CLL vs M-CLL samples. (B) Volcano plot of basal protein phosphorylation in untreated UM-CLL cells vs M-CLL. (C) pS:pT:pY ratio of all isolated p-peptides in untreated UM-CLL cells vs M-CLL. (D) pS:pT:pY ratio of all differentially regulated p-peptides in untreated UM-CLL cells vs M-CLL. (E) Heat map representing basic phosphorylation levels of the most relevant regulated proteins in untreated M-CLL and UM-CLL cells (cell migration and motility). (F) pS:pT:pY ratios in M-CLL and UM-CLL upon BCR stimulation compared with native cells (left columns), ibrutinib treatment compared with native (nat.) cells (middle columns), and a combination of BCR stimulation and ibrutinib treatment compared with BCR stimulation alone (right columns). (G) Number of differentially regulated p-peptides in UM-CLL and M-CLL upon BCR stimulation compared with native, with corresponding volcano plot. (H) Number of differentially regulated p-peptides in UM-CLL and M-CLL upon ibrutinib treatment compared with native cells and corresponding volcano plot. (I) Number of differentially regulated p-peptides upon ibrutinib treatment plus BCR stimulation compared with BCR stimulation only with corresponding volcano plot.
p-proteome analyses reveal distinct phosphorylation patterns in UM-CLL and M-CLL. (A) Number of differentially regulated p-peptides in untreated UM-CLL vs M-CLL samples. (B) Volcano plot of basal protein phosphorylation in untreated UM-CLL cells vs M-CLL. (C) pS:pT:pY ratio of all isolated p-peptides in untreated UM-CLL cells vs M-CLL. (D) pS:pT:pY ratio of all differentially regulated p-peptides in untreated UM-CLL cells vs M-CLL. (E) Heat map representing basic phosphorylation levels of the most relevant regulated proteins in untreated M-CLL and UM-CLL cells (cell migration and motility). (F) pS:pT:pY ratios in M-CLL and UM-CLL upon BCR stimulation compared with native cells (left columns), ibrutinib treatment compared with native (nat.) cells (middle columns), and a combination of BCR stimulation and ibrutinib treatment compared with BCR stimulation alone (right columns). (G) Number of differentially regulated p-peptides in UM-CLL and M-CLL upon BCR stimulation compared with native, with corresponding volcano plot. (H) Number of differentially regulated p-peptides in UM-CLL and M-CLL upon ibrutinib treatment compared with native cells and corresponding volcano plot. (I) Number of differentially regulated p-peptides upon ibrutinib treatment plus BCR stimulation compared with BCR stimulation only with corresponding volcano plot.
Phosphorylation is a common posttranslational modification that enables regulation, function, and conformation of the protein. It occurs most frequently on serine residues, followed by threonine and tyrosine residues.61,62 Comparing the ratio of phosphoserine (pS) to phosphothreonine (pT) to phosphotyrosine (pY) of all detected peptides, M-CLL and UM-CLL (pS:pT:pY, 89:10:1) cells exhibited a physiological ratio (Figure 4C).62 However, looking at the pS:pT:pY ratio of all differentially regulated peptides, a significant shift toward pY could be detected (pS:pT:pY, 83:8:9) (Figure 4D). Further analyses revealed that phosphopeptides with a distinct phosphorylation pattern predominantly clustered in pathways associated with migration and motility (Figure 4E). Interestingly, similar to the results from proteomic analysis, phosphorylation of MARCKS showed significant differences between UM-CLL and M-CLL. Phosphorylation was detected at 3 distinct sites (S101, T150, and S170) (Figure 4E).
BCR stimulation increases phosphorylation independent of CLL subtype
Stimuli from the tumor microenvironment (TME) are important for survival and proliferation of CLL cells, and BCR signaling plays a pivotal role in mediating these stimuli.63 However, the effect of BCR stimulation on the p-proteome has only been investigated for individual proteins so far.64 In our analyses, BCR stimulation led to an increase of phosphorylation in M-CLL (98 of 106 proteins) and UM-CLL (112 of 129 proteins) (Figure 4G). Phosphorylation of proteins across all relevant pathways was affected by BCR stimulation (supplemental Table 3A). For instance, we detected increased phosphorylation of proteins associated with BCR signaling in both UM-CLL and M-CLL cells after BCR stimulation (supplemental Table 3A), for example, caspase recruitment domain-containing protein 11 (CARD11), phosphoinositide 3-kinase adapter protein 1 (PI3KAP1), docking protein 3 (DOK3), or inositol polyphosphate-5-phosphatase D (INPP5D).
Ibrutinib treatment induces differential signaling depending on IGHV status
Next, we investigated the impact of ibrutinib on the p-proteome. We found that the effect of ibrutinib was different depending on IGHV mutational status. Whereas hyperphosphorylation in UM-CLL cells was reduced to baseline levels after ibrutinib treatment, with 40 of 44 proteins showing a decrease in phosphorylation (40 of 44), we saw a relative increase of phosphorylation in M-CLL cells (34 of 43) upon ibrutinib treatment (Figure 4H; supplemental Table 3B). Most of the proteins regulated by ibrutinib were associated with BCR signaling (eg, LYN, Intersectin 2 [ITSN2], tyrosine-protein phosphatase nonreceptor type 6 [PTPN6], PTPN11, Cofilin 1 [CFL1], or platelet endothelial cell adhesion molecule [PECAM1]).
Regarding the pS:pT:pY ratio, ibrutinib treatment induced a shift of the ratio only in UM-CLL. In M-CLL, only a slight shift toward pY could be observed (pS:pT:pY, 78:7:15), whereas 61% of all regulated phosphopeptides in UM-CLL cells were peptides with a phosphotyrosine (pS:pT:pY, 30:9:61) (Figure 4F; supplemental Table 3B).
In a next step, we investigated whether ibrutinib can reverse the hyperphosphorylation induced by BCR stimulation, which was found in both M-CLL and UM-CLL. We found that combining ibrutinib and BCR stimulation induced a decrease in phosphorylation in 43 of 44 proteins in UM-CLL compared with BCR stimulation only, whereas, in M-CLL, the ratio between upreulation and downregulation of phosphorylation (7 of 13 vs 6 of 13 proteins) was almost balanced with no striking differences regarding the pS:pT:pY ratio (Figure 4F,I; supplemental Table 3C).
MARCKS is expressed at significantly higher levels in M-CLL
A protein that specifically attracted our attention during our analysis was MARCKS, which is known to play a role in invasiveness and metastasis in various solid tumors.16-18 Interestingly, MARCKS was recently described to be relevant for the nanoclustering of the BCR and thereby for the lateral mobility of the BCR with influence on both tonic and chronic active BCR signaling.11 However, although MARCKS expression in CLL and MCL has been reported, its functional role has not yet been elucidated.12 In their report, Vargova et al also describe that expression of MARCKS is significantly lower in CLL cells compared with normal B cells. We confirmed this with data from a previously published transcriptome analysis on CLL and normal B cells.65 MARCKS expression was higher in normal naive, IgG memory, and IgM memory B cells than in M-CLL and UM-CLL cells (Figure 5A).
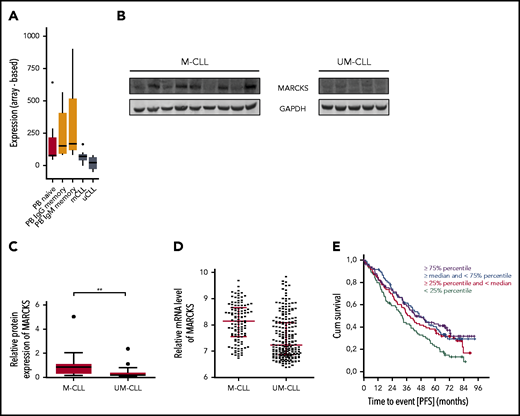
MARCKS in CLL and normal B cells. (A) MARCKS transcript expression signal intensity of normal naive, IgG memory, and IgM memory B cells isolated from peripheral blood (PB) and M-CLL and UM-CLL tumor cells from 5 donors each. (B-C) Immunoblot for MARCKS expression in M-CLL and UM-CLL (B) and corresponding densitometric analysis (C) of n = 36 patients. (D) Relative messenger RNA (mRNA) expression of MARCKS in M-CLL and UM-CLL in 337 patients. (E) Correlation of MARCKS expression with PFS of 337 patients with CLL treated with FC or FCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
MARCKS in CLL and normal B cells. (A) MARCKS transcript expression signal intensity of normal naive, IgG memory, and IgM memory B cells isolated from peripheral blood (PB) and M-CLL and UM-CLL tumor cells from 5 donors each. (B-C) Immunoblot for MARCKS expression in M-CLL and UM-CLL (B) and corresponding densitometric analysis (C) of n = 36 patients. (D) Relative messenger RNA (mRNA) expression of MARCKS in M-CLL and UM-CLL in 337 patients. (E) Correlation of MARCKS expression with PFS of 337 patients with CLL treated with FC or FCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Our MS analyses revealed phosphorylation of MARCKS at different phosphosites and a striking expression difference in UM-CLL and M-CLL samples, with MARCKS being upregulated in M-CLL (Figures 3E and 4E; supplemental Figure 2). To validate this finding, we analyzed expression of MARCKS by immunoblot in 36 different patients with CLL (16 UM-CLL, 20 M-CLL) and could confirm our results from the proteome analysis, showing significantly higher expression of MARCKS in M-CLL (Figure 5B-C). We validated this finding on the transcriptional level in a cohort of 337 patients with CLL (false discovery rate <0.001; fold change, 1.5; Figure 5D).
MARCKS regulates migration and AKT signaling
To investigate the cellular functions of MARCKS, we used CRISPR/Cas9 to generate MARCKS KO OSU-CLL (OSU KO) cells (Figure 6A). OSU KO and the control cells (OSU ctrl) were treated with CXCL12 (SDF1) and migration was investigated after 4 hours. Loss of MARCKS led to significantly increased migration of OSU-CLL cells (Figure 6B). To validate this finding, we generated MARCKS knockdown by siRNA in OSU-CLL cells. In line with our previous results, migration toward CXCL12 was also increased in the knockdown cells (supplemental Figure 3A).
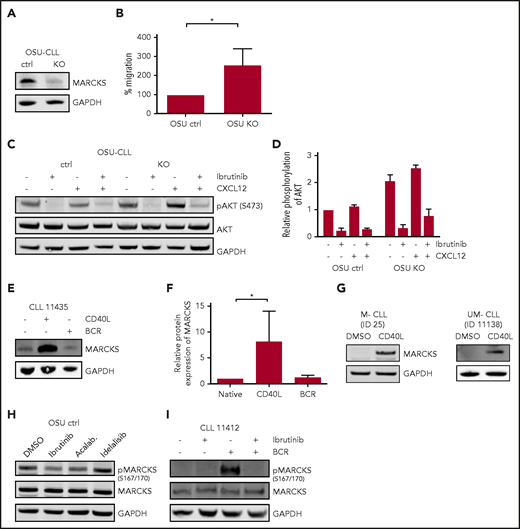
Regulation and functional relevance of MARCKS. (A) Immunoblot for MARCKS in CRISPR /Cas9 OSU KO and respective OSU ctrl cells. (B) Migration (in percentage input control) toward CXCL12 in OSU KO and OSU ctrl normalized to OSU ctrl. (C) Immunoblot for AKT phosphorylation (S473) in OSU KO and OSU ctrl after treatment with ibrutinib (1 hour) and/or CXCL12 (2 minutes) and (D) corresponding densitometric analysis. (E) Immunoblot for MARCKS expression upon CD40L or BCR stimulation in a patient with CLL (blot representative of 7 different patients) with (F) densitometric analysis of n = 7 patients. (G) Induction of MARCKS in M-CLL and UM-CLL by CD40L stimulation. Blots are representative of 5 patients, respectively. (H) Immunoblot for MARCKS phosphorylation (S167/170) in OSU-CLL cells upon treatment with BTK inhibitors (1 µM, 60 minutes) ibrutinib and acalabrutinib (Acalab.) and PI3K inhibitor idelalisib (1 µM, 60 minutes). Blot is representative of n = 3 blots. (I) Immunoblot showing MARCKS phosphorylation (S167/170) upon ibrutinib treatment and/or BCR stimulation in a sample of a patient with CLL (blot representative of 4 different patients). DMSO, dimethyl sulfoxide.
Regulation and functional relevance of MARCKS. (A) Immunoblot for MARCKS in CRISPR /Cas9 OSU KO and respective OSU ctrl cells. (B) Migration (in percentage input control) toward CXCL12 in OSU KO and OSU ctrl normalized to OSU ctrl. (C) Immunoblot for AKT phosphorylation (S473) in OSU KO and OSU ctrl after treatment with ibrutinib (1 hour) and/or CXCL12 (2 minutes) and (D) corresponding densitometric analysis. (E) Immunoblot for MARCKS expression upon CD40L or BCR stimulation in a patient with CLL (blot representative of 7 different patients) with (F) densitometric analysis of n = 7 patients. (G) Induction of MARCKS in M-CLL and UM-CLL by CD40L stimulation. Blots are representative of 5 patients, respectively. (H) Immunoblot for MARCKS phosphorylation (S167/170) in OSU-CLL cells upon treatment with BTK inhibitors (1 µM, 60 minutes) ibrutinib and acalabrutinib (Acalab.) and PI3K inhibitor idelalisib (1 µM, 60 minutes). Blot is representative of n = 3 blots. (I) Immunoblot showing MARCKS phosphorylation (S167/170) upon ibrutinib treatment and/or BCR stimulation in a sample of a patient with CLL (blot representative of 4 different patients). DMSO, dimethyl sulfoxide.
Unphosphorylated MARCKS resides at the cell membrane and inhibits downstream signaling, for example, PI3K/AKT signaling by sequestration of PIP2.13-15,66 Phosphorylation of membrane-bound MARCKS by PKC leads to translocation of MARCKS to the cytosol and release of PIP2.12 Therefore, we investigated phosphorylation of proteins in these pathways in OSU KO cells. We found that KO of MARCKS led to increased phosphorylation of AKT (S473) after CXCL12 stimulation compared with WT MARCKS, indicating an inhibitory effect of MARCKS on AKT signaling (Figure 6C-D).
Xu et al have recently shown that MARCKS expression reduces intensity of BCR signaling.11 CLL cells from patients with unmutated IGHV exhibit stronger BCR signaling in comparison with patients with mutated IGHV. However, the activation level of the BCR pathway within the UM-CLL cells varies. To characterize the role of MARCKS as a fine-tuning element of the BCR independent of IGHV status, we investigated BCR signaling in UM-CLL samples with low and high MARCKS expression, as determined by qPCR. Phosphorylation of AKT and ERK1/2 was investigated by immunoblot. Phosphorylation of AKT was significantly stronger (P = .016) in patients with low MARCKS expression. Phosphorylation of ERK was also enhanced in patients with low MARCKS (supplemental Figure 3B-D).
MARCKS is regulated by the TME and can be targeted by BTK inhibitors but not by idelalisib
As MARCKS seems to play a significant role in central signaling pathways, we wondered whether and how MARCKS is regulated by the TME in CLL cells, and whether its phosphorylation can be targeted by BTK or PI3K inhibitors. Therefore, we investigated the influence of typical stimuli from the TME (CD40:CD40L interaction and stimulation of the BCR) on expression and regulation of MARCKS. T-cell–mediated CD40 stimulation induces proliferation of CLL cells in the lymph nodes.67-69 Interestingly, only stimulation with CD40L-expressing feeder cells led to an induction in total MARCKS protein expression in CLL cells, whereas BCR stimulation did not affect total protein level (Figure 6E-F). MARCKS expression could be induced both in UM-CLL (n = 5) and M-CLL (n = 5) (Figure 6G), but as intensity of MARCKS was too low for accurate quantification in some unstimulated samples, densitometric analysis was not performed. Intriguingly, comparing MARCKS expression in different compartments, expression was lower in peripheral blood compared with nodal tissue (supplemental Figure 4). In contrast to total protein expression, MARCKS phosphorylation (S167/170) in CLL cells is only induced by BCR stimulation but not CD40-to-CD40L interaction (Figure 6I). Furthermore, we found that both BTK inhibitors ibrutinib and acalabrutinib led to a decrease in phosphorylation (S167/170) in OSU-CLL cells. PI3K inhibitor idelalisib, which acts upstream of MARCKS, did not affect MARCKS phosphorylation (Figure 6H).
MARCKS expression correlates with differential response to BTK inhibitor therapy
To validate the clinical relevance of our in vitro observations, we analyzed MARCKS expression in 2 cohorts of patients treated with (immuno)chemotherapy or acalabrutinib, respectively.
In a cohort of 337 CLL patients enrolled in the CLL 8 cohort, we found that there is shorter progression-free survival (PFS) in patients with low MARCKS irrespective of the IGHV status. In detail, in a Cox regression calculation, low MARCKS was prognostically significant independent of the treatment arm (fludarabine, cyclophosphamide, and rituximab [FCR] or fludarabine and cyclophosphamide [FC] and UM-CLL vs M-CLL). However, 1 of the quartile curves (50% to 75% expression) was not statistically significant in this calculation (Figure 5E). The observation time was 7 years.
We were able to demonstrate the clinical relevance of MARCKS expression for patients treated with BTK inhibitors. We investigated the relationship between MARCKS expression and the response to acalabrutinib therapy in a recently published cohort of patients with CLL.48 As expected, MARCKS messenger RNA expression measured by RNA-seq was widely variable with highest expression in 2 patients with M-CLL; it was generally lower in 18 patients with UM-CLL (supplemental Figure 5). In patients with below median MARCKS expression compared with those with median or higher expression, treatment-induced lymphocytosis was more prominent and absolute lymphocyte count (ALC) was significantly higher at the end of cycle 1 (P = .019) and cycle 2 (P = .048) (Figure 7A-B), whereas the pretreatment ALC was not different between the 2 groups. With continued therapy, lymphocytosis resolved in both groups. Conversely, the decrease in nodal disease was more pronounced in patients with below median MARCKS expression (P = .037; Figure 7C-D). All patients in both groups achieved an objective response, albeit with slower onset of response in patients with below median MARCKS expression. As the cohort was unbalanced (18 UM-CLL, 2 M-CLL), the same analysis was performed for UM-CLL samples only and showed comparable results. Treatment-induced lymphocytosis was higher in patients with UM-CLL that are below median MARCKS expression (supplemental Figure 6) and the decrease in nodal size was also more pronounced (supplemental Figure 7). Looking at MARCKS expression in circulating tumor cells during treatment with acalabrutinib, we found that it decreased after the first cycle of acalabrutinib, coinciding with the period of treatment-induced lymphocytosis, then increased again by the end of cycle 6 (Figure 7E).
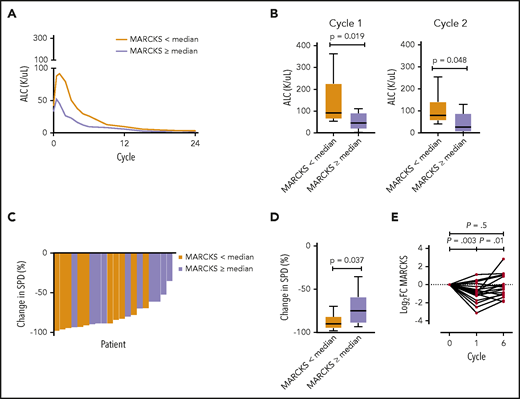
Correlation of MARCKS expression with response to BTK inhibition. (A) Acalabrutinib-induced lymphocytosis dependent on MARCKS expression determined by RNA-seq (n = 20: 18 UM-CLL, 2 M-CLL). (B) ALC dependent on MARCKS expression after cycle 1 and cycle 2 of acalabrutinib treatment. (C-D) Acalabrutinib induced change in nodal disease in patients with below and above median MARCKS expression. (E) Expression of MARCKS during 6 cycles of treatment with acalabrutinib in 20 patients. Log2 fold change from baseline is shown for each patient (mean log2 fold change, −0.8675 after cycle 1). Paired Student t test. SPD, sum of the products of lymph node diameters.
Correlation of MARCKS expression with response to BTK inhibition. (A) Acalabrutinib-induced lymphocytosis dependent on MARCKS expression determined by RNA-seq (n = 20: 18 UM-CLL, 2 M-CLL). (B) ALC dependent on MARCKS expression after cycle 1 and cycle 2 of acalabrutinib treatment. (C-D) Acalabrutinib induced change in nodal disease in patients with below and above median MARCKS expression. (E) Expression of MARCKS during 6 cycles of treatment with acalabrutinib in 20 patients. Log2 fold change from baseline is shown for each patient (mean log2 fold change, −0.8675 after cycle 1). Paired Student t test. SPD, sum of the products of lymph node diameters.
\Furthermore, we analyzed MARCKS expression in 5 CLL patients (1 M-CLL, 4 UM-CLL) before ibrutinib treatment and at progression. Interestingly, MARCKS expression was higher at progression compared with baseline samples (supplemental Figure 8). In line with the data on MARCKS expression during acalabrutinib treatment, expression of MARCKS dropped after initiation of ibrutinib therapy in 4 of 5 patients and returned to or above baseline levels during further treatment or until disease progression (supplemental Figure 8).
Discussion
To understand the efficacy of BTK inhibitors in patients with CLL that have unmutated IGHV (UM-CLL), we investigated the proteome and p-proteome of primary CLL cells.3,70-72 Prior reports focusing on selected proteins identified differential activation of pathways in UM-CLL cells, leading to a survival advantage.5,9,21,22 We selected a different approach studying differences in the (p-)proteome in M-CLL vs UM-CLL. Despite starting with a limited number of samples (n = 3 for UM-CLL and M-CLL, respectively), we were able to identify relevant differences that were then reproduced in a larger number of samples and on a functional level.
In essence, our data revealed differences of the p-proteome, with >83% of differentially phosphorylated proteins showing higher basal phosphorylation in UM-CLL. Interestingly, higher levels of phosphorylation were observed on proteins involved in migration and motility, which could be targeted by ibrutinib. Comparing the proteome of M-CLL and UM-CLL, differentially expressed proteins also accumulated in pathways associated with migration/motility and cell survival, in line with a report of Eagle et al in which MARCKS-related protein was detected but not MARCKS itself.28 Of note, our data also highlighted differential expression of proteins involved in cell metabolism (AK1, COX5B) and oxidative stress (NRF2 signaling). These proteins have not been described in CLL before and might be interesting targets for future studies.
The protein MARCKS attracted our specific attention during our analyses due to striking differences in protein expression and phosphorylation status. MARCKS, a PKC substrate, sequesters PIP2 and thereby regulates central signaling pathways like PI3K/AKT signaling.13-15 Upon phosphorylation, membrane-bound MARCKS translocates to the cytoplasm and releases PIP2. PIP2 can then be phosphorylated by PI3K, which is essential for recruitment of AKT to the membrane and for subsequent activation.73 Data from these studies, showing an inhibitory effect of MARCKS on downstream signaling,15 could be confirmed in our experiments. Looking at MARCKS biology, it becomes clear that cells with low MARCKS expression, like UM-CLL cells, most likely have higher levels of free PIP2 due to missing sequestration with subsequent induction of downstream signaling.13,14,74 Although we do not show PIP2 assays, we were able to show that regulation of PIP2 targets like AKT were dependent on MARCKS expression in OSU cells. AKT signaling upon CXCL12 treatment was significantly higher in MARCKS KO cells. Furthermore, migration toward CXCL12, which is known to be PIP2 dependent,75 was higher in MARCKS KO cells.
Increased activity of pathways downstream of the BCR in cells with low MARCKS expression might explain the trend of worse PFS we saw in patients treated with FC or FCR.
Given the distinct expression of MARCKS in UM-CLL and M-CLL, low MARCKS expression could lead to increased chemotaxis of UM-CLL cells and increased downstream signaling. Indeed, Eagle et al have shown that UM-CLL cells are preferentially retained in the lymphatic tissues.28
Xu et al associated MARCKS with BCR clustering.11 They found, that membrane-tethered MARCKS increases BCR lateral mobility, and thus decreases BCR nanoclustering by disturbing the interaction between cortical F-actin and the inner leaflet of the plasma membrane. This leads to suppression of the strength of both tonic and chronic active BCR signaling.11 In line with their data, our results suggest that MARCKS acts as a fine-tuning element of the BCR via PIP2 interaction. Activation of BCR signaling leads to phosphorylation of MARCKS, resulting in higher amounts of unsequestered PIP2 and enhanced downstream signaling. Inhibition of BTK leads to the opposite.
Naturally, analyses performed in cell lines have their limitations. However, we only used the cell line to characterize the cellular function of a single protein and were able to transfer these results to in vivo data. Our in vitro functional data on MARCKS is supported by in vivo data from patients with CLL treated with acalabrutinib.
Indeed, we were able to show that response to acalabrutinib correlates with MARCKS expression. Experimentally, knockdown of MARCKS enhanced cell motility and, in patients treated with acalabrutinib, below median MARCKS expression was associated with increased treatment-induced lymphocytosis and a stronger reduction in lymph node size. Patients with M-CLL treated with BTK inhibitors generally have more prolonged lymphocytosis than those with UM-CLL. However, onset and peak of the treatment-induced lymphocytosis in UM-CLL occur within days of starting therapy, whereas the peak in M-CLL is only reached after months on treatment. This interpretation is consistent with prior data demonstrating rapid release of CLL cells from nodal stations upon initiation of BTK inhibitor therapy, concomitant with inhibition of cellular migration and adhesion.76-79 As the lymphocytosis results from exit of cells from the lymphoid tissues, faster onset and an earlier peak are consistent with higher motility, which is supported by the transient decrease in MARCKS expression in circulating tumor cells during the early treatment period with acalabrutinib. Intriguingly, in patients with progressive disease under ibrutinib treatment, expression of MARCKS was upregulated.
As 18 of the 20 patients treated with acalabrutinib had UM-CLL, our observations support the concept that MARCKS expression is a biomarker for the response to BTK inhibitor therapy independent of IGHV status. Moreover, MARCKS seems a functionally relevant modulator of BCR signaling in CLL.
Acknowledgments
The authors thank Ellen Kendall for bioinformatic support. The authors are indebted to their patients, who provided primary material.
This work was supported by the Deutsche Forschungsgemeinschaft (KFO-286 [L.P.F. and M.H.], SFB1074 subproject B1 [S.S.]), and the Jose Carreras Stiftung (DJCLS-10R/2018 [L.P.F.]). L.P.F. and M.H. were supported by a Gilead Sciences research grant. A.W. and C.S. were supported by the intramural program of the National Institutes of Health, National Heart, Lung, and Blood Institute (HL002346-15). R.P.Z. is grateful to Genome Canada GTP platform funding for operations and technology development (264PRO) and thanks the Ministry of Science and Higher Education of the Russian Federation (MegaGrant, Agreement with Skolkovo Institute of Science and Technology [075-10-2019-083]).
Authorship
Contribution: R.P.Z. and L.P.F. conducted the research plan; L.B., V.B., C.D., O.M., M.O., J.C., C.U., L.V., D.T., S.L., M.S., and R.P.Z. performed experiments; K.F., A.M.F., B.E., C.S., and A.W. conducted the clinical trials; L.B., V.B., C.D., J.B., S.R., A.d.P.G., M.F.H., S.L., C.M.W., A.S., S.S., K.F., M.H., R.P.Z., M.S., E.T., A.W., C.S., and L.P.F. analyzed the data; S.L., A.S., and S.S. provided material, reagents, and equipment; and L.B., V.B., R.P.Z., A.W., and L.P.F. wrote the paper.
Conflict-of-interest disclosure: K.F. received AbbVie honoraria and Roche travel grants and honoraria. L.P.F. received AbbVie honoraria and travel grants, Roche honoraria, and research grants from AbbVie, Roche, and Gilead. A.W. received research support from Pharmacyclics LLC, an AbbVie Company; Acerta LLC, a member of AstraZeneca Group; Merck; Nurix; Verastem; and Genmab. C.S. received research support from Genmab. The remaining authors declare no competing financial interests.
Correspondence: Lukas P. Frenzel, Department I of Internal Medicine, University Hospital Cologne, Kerpener Str 62, 50937 Cologne, Germany; e-mail: lukas.frenzel@uk-koeln.de.
The data/mass spectrometry data reported in this article have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (data set identifier PXD016421). Data retrieved from HG U133 2.0 Plus GeneChip array analyses were submitted to the Gene Expression Omnibus (GEO) database (accession number GSE36907). The RNA-seq data from acalabrutinib-treated patients reported in this article have been deposited in the GEO data repository (accession number GSE136634).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal