

In this issue of Blood, 1 compare a proteomic screen of chronic lymphocytic leukemia (CLL) cells isolated from patients with mutated immunoglobulin heavy-chain variable region (IGHV) (M-CLL) to unmutated IGHV (UM-CLL). Among differentially expressed proteins, they found myristoylated alanine-rich C-kinase substrate (MARCKS) to be highly expressed in M-CLL patients, and low in patients with UM-CLL. They convincingly link the expression and phosphorylation of MARCKS to CLL cell migration (ie, key CLL signaling pathways, especially CXCR4 and B-cell receptor [BCR] signaling). They also corroborate the findings in samples from patients receiving treatment with the Bruton tyrosine kinase (BTK) inhibitor acalabrutinib.
MARCKS initially was described as a phosphoprotein highly expressed in neuronal tissues; the importance of MARCKS for neural development was further emphasized by the phenotype of MARCKS-null mice, which is embryonically lethal due to severe neural defects.2 Subsequently, MARCKS was characterized as a multifunctional regulatory protein involved in multiple signaling pathways. It tethers phosphatidylinositol 4,5-bisphosphate (PIP2) to the cell membrane, and binds calmodulin and F-actin, thereby regulating cell motility and the function of the cytoskeleton.3,4 MARCKS, a specific substrate of protein kinase C (PKC), relocates from the plasma membrane to the cytoplasm, once MARCKS becomes phosphorylated (see figure). Besides its role in cell migration, MARCKS has been linked to B-cell survival and proliferation by coregulating the function of BCR. Xu et al reported that MARKS can differentially regulate BCR signaling and the fate of B cells, depending on localization, expression, and activation of MARCKS. High MARCKS expression and membrane-tethered MARCKS reduces BCR signaling by disturbing BCR clustering in response to antigen. Activated cytoplasmatic MARCKS, on the other hand, allows for robust BCR signaling, resulting in proliferation of malignant and primary B cells in vitro and in vivo.5 Once MARCKS is activated, it dissociates from the cell membrane and thereby makes PIP2 more accessible for activation through the phosphatidylinositol 3-kinase (PI3K) pathway. In CLL cells, MARCKS expression can be induced by phorbol 12-myristate 13acetate (PMA), and expression levels generally are lower than in other B-cell malignancies.6 Beckmann et al demonstrated in their current report that the expression of MARCKS is decidedly different between samples from patients with mutated IGHV (M-CLL, MARCKS highly expressed) vs unmutated IGHV (UM-CLL, MARCKS low).1 This is of great interest given the profound differences in disease biology and response to treatment in these patient subsets. Typically, patients with M-CLL have more indolent CLL, and their BCRs are less responsive to stimulation, whereas the B-cell clones from patients with UM-CLL have robust BCR signaling and proliferate faster. Therefore UM-CLL patients generally have more active disease and a shorter time to requiring treatment.7
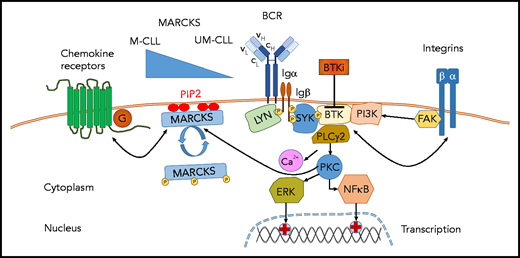
MARCKS modulates CLL cell signaling. MARCKS tethers PIP2 to the cell membrane and binds Ca2+/calmodulin and F-actin, thereby regulating cell motility and the function of the cytoskeleton. By hindering BCR clustering and capturing PIP2, thereby limiting availability of PIP2 for PI3K-related signaling, membranous MARCKS tones down signaling of the BCR and chemokine receptors. In this issue of Blood, Beckmann et al demonstrate that M-CLL patients have higher MARCKS levels, and UM-CLL patients have lower MARCKS levels. This differential MARCKS expression correlates with higher or lower capacity for BCR and chemokine receptor signaling, respectively. These preclinical findings are translated into the clinical situation in CLL patients treated with the BTK inhibitor acalabrutinib. Acalabrutinib, as seen with all BTK inhibitors, induces redistribution lymphocytosis due to the mobilization of lymph node–resident CLL cells into the peripheral blood. This redistribution lymphocytosis is more pronounced in patients with low MARCKS and UM-CLL (and high signaling activity/dependency) and less pronounced in patients with high MARCKS (lower signaling activity/dependency). The signaling modulation by MARCKS is further regulated by its activation/phosphorylation status; phosphorylation of MARCKS (eg, from enhanced BCR signaling) results in relocation of MARCKS from the membrane into the cytoplasm, thereby fostering BCR clustering and enhanced access to PIP2 and strengthening BCR- and migration-related signaling, preferentially in UM-CLL. BTKi, BTK inhibitor; Ig, immunoglobulin.
MARCKS modulates CLL cell signaling. MARCKS tethers PIP2 to the cell membrane and binds Ca2+/calmodulin and F-actin, thereby regulating cell motility and the function of the cytoskeleton. By hindering BCR clustering and capturing PIP2, thereby limiting availability of PIP2 for PI3K-related signaling, membranous MARCKS tones down signaling of the BCR and chemokine receptors. In this issue of Blood, Beckmann et al demonstrate that M-CLL patients have higher MARCKS levels, and UM-CLL patients have lower MARCKS levels. This differential MARCKS expression correlates with higher or lower capacity for BCR and chemokine receptor signaling, respectively. These preclinical findings are translated into the clinical situation in CLL patients treated with the BTK inhibitor acalabrutinib. Acalabrutinib, as seen with all BTK inhibitors, induces redistribution lymphocytosis due to the mobilization of lymph node–resident CLL cells into the peripheral blood. This redistribution lymphocytosis is more pronounced in patients with low MARCKS and UM-CLL (and high signaling activity/dependency) and less pronounced in patients with high MARCKS (lower signaling activity/dependency). The signaling modulation by MARCKS is further regulated by its activation/phosphorylation status; phosphorylation of MARCKS (eg, from enhanced BCR signaling) results in relocation of MARCKS from the membrane into the cytoplasm, thereby fostering BCR clustering and enhanced access to PIP2 and strengthening BCR- and migration-related signaling, preferentially in UM-CLL. BTKi, BTK inhibitor; Ig, immunoglobulin.
Before the introduction of BTK inhibitors (ibrutinib, acalabrutinib, and zanubrutinib) in CLL therapy, patients with M-CLL generally had a better prognosis because they responded well to chemoimmunotherapy. Conversely, UM-CLL patients have shorter remissions and inferior survival with chemotherapy-based treatment. However, when targeting BCR signaling with ibrutinib, the outcome of patients in both CLL subgroups is virtually identical in long-term outcome data with single-agent ibrutinib in previously untreated CLL patients.8 Ergo, BTK inhibitors can overcome the negative prognostic impact of U-CLL by targeting a key signaling molecule, BTK, which has a prominent role in BCR signaling and the signaling of other surface receptors related to B-cell migration and homing (ie, chemokine receptors and adhesion molecules).9 Consequently, when blocking BTK with these new agents, BCR signaling and BCR-associated CLL cell survival and proliferation are blocked. Additionally, the function of the tissue-homing receptors is disturbed. Patients experience mobilization of CLL cells from the tissue compartments into the peripheral blood, resulting in a transient surge in CLL cells in the peripheral blood.10 The degree and kinetics of redistribution lymphocytosis, and the degree and kinetics of resolution of lymphadenopathy, mirror activity and dependency of each CLL clone on signaling through the BCR and tissue-homing receptors. As shown by the investigators, levels of MARCKS expression in CLL cells from acalabrutinib-treated patients correlate quite well with these signs of BTK inhibitor–induced CLL cell redistribution. Patients with high expression of MARCKS, enriched in the M-CLL subset, have less complete resolution of lymphadenopathy and lower levels of redistribution lymphocytosis, whereas patients with low MARCKS experience higher levels of lymphocytosis and more complete resolution of enlarged lymph nodes.
This study provides fascinating new molecular insight into the regulation of signaling of BCR and tissue-homing receptors, demonstrating a previously unrecognized important role for MARCKS in these key regulatory disease pathways in CLL. At the same time, this study also provides new insight into the mechanism of action of BTK inhibitors, highlighting the involvement of MARCKS in fine-tuning responses to the BTK inhibitors, the new cornerstone of CLL therapy.
Conflict-of-interest disclosure: J.A.B. receives research funding from Pharmacyclics, BeiGene, and AstraZeneca and honoraria from Janssen.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal