

In this issue of Blood, 1 show that pediatric acute myeloid leukemia (AML) cells form close connections with mesenchymal stromal cells (MSCs) to reprogram them and that these cell–cell contacts serve as a vulnerability, which can be exploited to improve AML treatments.
Healthy hematopoietic cells, as well as their malignant counterparts, reside in a highly complex and dynamic bone marrow (BM) microenvironment formed by numerous cell types, including MSCs, which collectively make up the BM niche.2 Bidirectional cross talk between hematopoietic and stromal compartments is crucial to maintenance of bone marrow homeostasis. However, this communication can be hijacked by the leukemic cells to facilitate immune evasion and chemoresistance. Although a possible role for BM stromal cells in blood malignancy was hypothesized in the 1990s,3 an understanding of molecular mechanisms explaining how BM niche alterations affect mutated and wild-type hematopoietic cells in the context of malignancy has remained elusive.
Studying the BM microenvironment, especially in patients, poses numerous challenges: (1) a large volume of BM aspirate is required because of the relatively small proportion of stromal cells within the overall BM; (2) in vitro expansion may introduce artifacts; and (3) new treatment protocols may be difficult to test in cell culture systems that do not recapitulate the spatial and temporal complexity of cell interactions in the BM. Considering these issues, it is not surprising that most of our knowledge addressing BM niche biology comes from mouse models and samples from adult malignancies. One of the most intriguing and unresolved questions in the field is whether a perturbed BM niche can initiate leukemogenesis or is rather a consequence of leukemia that contributes to disease progression. Several studies have shown that niche-specific deletion of Rarg, Dicer1, Ikba, or Rb1 predisposes to myeloproliferative neoplasm-like disease in mice.4 Moreover, mice with constitutively active β-catenin or an activating mutation in Ptpn11 in niche cells develop AML and juvenile myelomonocytic leukemia-like disorder, respectively.4 Despite the results from animal models arguing that BM niche lesions can initiate hematologic malignancies, the same phenomenon remains to be determined in human patients.
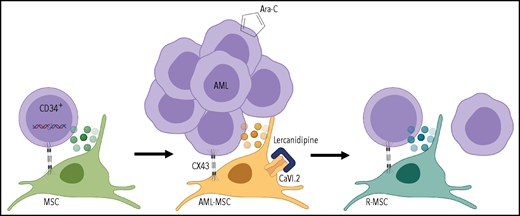
Mutations occur in CD34+ progenitors, leading to oncogenic transformation; AML cells reprogram the MSC transcriptome and secretome through CX43-mediated cell contact and create an onco-niche (AML-MSC). Dual targeting of MSCs with calcium channel (CaVI.2) blocker lercanidipine and AML cells with standard chemotherapeutic Ara-C reduces leukemia burden and restores healthy features of MSCs during the remission (R-MSC). Figure created with BioRender.com.
Mutations occur in CD34+ progenitors, leading to oncogenic transformation; AML cells reprogram the MSC transcriptome and secretome through CX43-mediated cell contact and create an onco-niche (AML-MSC). Dual targeting of MSCs with calcium channel (CaVI.2) blocker lercanidipine and AML cells with standard chemotherapeutic Ara-C reduces leukemia burden and restores healthy features of MSCs during the remission (R-MSC). Figure created with BioRender.com.
On the other hand, evidence from mouse models and human specimens suggests that malignant blood cells can reprogram MSCs to create a safe harbor that protects from cytotoxic therapy, immune surveillance, and nurtures cancer progression.5,6 Multiple studies have demonstrated that malignant cells establish close interactions with MSCs through direct cell–cell contact, tunneling nanotubes, or secreted vesicles to alter stromal cell transcriptomes and secretomes, which lead to activation of survival pathways, protection from excessive reactive oxygen species, and metabolic reprogramming of transformed cells.4,7 The so-called “onco-niche” exerts its pathogenic functions through increased secretion of proinflammatory cytokines (interleukin-6, interleukin-1β, or tumor necrosis factor α) and AML survival factors (GAS6), reduced production of hematopoietic stem cell–supporting factors (Cxcl12, stem cell factor, insulin-like growth factor 1), and excessive immunosuppression of T cells.4,5,8,9 Of note, profiling of the stromal compartment from patients with myelodysplastic syndrome (MDS) or AML suggests that MSC alterations, especially cytokine profile, can vary depending on disease stage and treatment. One cannot help but wonder whether there may be an opportunity for therapeutic intervention targeting not only mutated blood cells themselves but also the BM niche. Interestingly, analysis of MSCs from adult MDS or AML patients revealed that stromal cells can also harbor chromosomal abnormalities, although different than ones identified in blasts.5,8 However, the functional consequences of MSC genetic changes are not currently clear.
Borella et al extend our understanding of the BM niche to pediatric malignancy. The authors confirmed altered secretome and transcriptome in the MSCs of AML patients. They demonstrated reprograming of MSCs by cocultures of AML blasts with healthy MSCs and showed that connexin 43 (CX43)-based gap junctions were involved in this process (see figure). Profiled MSCs from a pediatric cohort did not exhibit cytogenetic or karyotypic changes found in the AML blasts. Intriguingly, MSCs reacquired healthy features during patient remission. A highlight of the study of Borella et al is a new 3-dimensional (3D) humanized biomimetic system simulating interaction of cells within the BM, which can be implemented in both in vitro and in vivo assays. This advanced coculture system potentiated the high-throughput chemical screen that identified lercanidipine, a calcium channel blocker approved to treat hypertension, as a potential new compound to target AML-MSCs. Using 3D cocultures of AML-MSCs and primary AML cells, the authors demonstrate that combining lercanidipine with compounds used in clinical care had a synergistic effect in decreasing leukemic cell proliferation. Most importantly, experiments using mice implanted with 3D scaffolds confirmed a reduced AML burden using the dual targeting approach (lercanidipine plus Ara-C) compared with single agents (see figure). Although undoubtedly important, the study has some caveats that should be considered. First, ex vivo expansion of patient MSCs was still required, potentially biasing transcriptome and secretome profiles or altering functional assay results. Second, leukemia burden in mice was followed for 5 weeks after treatment discontinuation, and further analysis is needed to determine durability of the dual targeting approach.
Considering that malignant cells can rewire MSCs transcriptional networks and reciprocally that perturbed BM niche cells can instigate persistent changes in the chromatin landscape of hematopoietic stem cells, it will be critical to define the molecular mechanisms underlying blood–niche communication.4,10 One might postulate a role for such interactions in leukemias that arise in transplant recipients where healthy transplanted cells encounter a niche damaged by primary disease and myeloablative treatments. Similarly, germline predisposition mutations, much more frequent in pediatric MDS/AML, may alter the niche, driving transformation or progression. How cross talk between MSCs and other niche components such as endothelial cells, osteoblasts, sympathetic nerves, and immune cells shapes the niche and affects malignant cells also remains to be determined. The current study extends our understanding of MSC reprograming in pediatric AML and provides proof-of-principle findings arguing that the BM niche is a promising therapeutic target in hematologic malignancies.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal