

Key Points
Ruxolitinib is clinically efficacious across various T-cell lymphoma subtypes, especially T-LGL.
Ruxolitinib activity is enriched among PTCL with JAK/STAT mutations or active signaling.

Abstract
Signaling through JAK1 and/or JAK2 is common among tumor and nontumor cells within peripheral T-cell lymphoma (PTCL). No oral therapies are approved for PTCL, and better treatments for relapsed/refractory disease are urgently needed. We conducted a phase 2 study of the JAK1/2 inhibitor ruxolitinib for patients with relapsed/refractory PTCL (n = 45) or mycosis fungoides (MF) (n = 7). Patients enrolled onto 1 of 3 biomarker-defined cohorts: (1) activating JAK and/or STAT mutations, (2) ≥30% pSTAT3 expression among tumor cells by immunohistochemistry, or (3) neither or insufficient tissue to assess. Patients received ruxolitinib 20 mg PO twice daily until progression and were assessed for response after cycles 2 and 5 and every 3 cycles thereafter. The primary endpoint was clinical benefit rate (CBR), defined as the combination of complete response, partial response (PR), and stable disease lasting at least 6 months. Only 1 of 7 patients with MF had CBR (ongoing PR > 18 months). CBR among the PTCL cases (n = 45) in cohorts 1, 2, and 3 were 53%, 45%, and 13% (cohorts 1 & 2 vs 3, P = .02), respectively. Eight patients had CBR > 12 months (5 ongoing), including 4 of 5 patients with T-cell large granular lymphocytic leukemia. In an exploratory analysis using multiplex immunofluorescence, expression of phosphorylated S6, a marker of PI3 kinase or mitogen-activated protein kinase activation, in <25% of tumor cells was associated with response to ruxolitinib (P = .05). Our findings indicate that ruxolitinib is active across various PTCL subtypes and support a precision therapy approach to JAK/STAT inhibition in patients with PTCL. This trial was registered at www.clincialtrials.gov as #NCT02974647.
Introduction
The peripheral T-cell lymphomas (PTCLs) and non-mycosis fungoides (non-MF) cutaneous T-cell lymphomas (CTCLs) represent a markedly heterogenous group of diseases characterized by relatively low cure rates with initial therapy and limited options for relapsed or refractory disease.1 Romidepsin, belinostat, pralatrexate, and brentuximab vedotin (BV) are US Food and Drug Administration-approved for these diseases and, with the exception of BV, response rates are disappointing (25% to 29%), progression-free survival is <4 months, and predictive biomarkers are lacking.2-5 BV, the anti-CD30 antibody drug conjugate, is highly active in CD30+ T-cell lymphomas (TCLs), particularly anaplastic large cell lymphoma (ALCL), and has been incorporated into front-line treatment, where it improved both progression-free survival and overall survival for patients with newly diagnosed CD30+ TCLs.6 Additional biomarker-driven therapeutics are desperately needed for all of the other TCLs.
The JAK/STAT pathway is activated in many TCL entities and therefore represents a potential therapeutic target. Activating JAK1, JAK2, JAK3, STAT3, or STAT5B mutations are observed in approximately 75% of cases of T-cell prolymphocytic leukemia (T-PLL),7 40% of T-cell large granular lymphocytic leukemia (T-LGL),8,9 and 33% of γδ–T-cell lymphomas (γδ-TCL).10 In addition, multiple cytokines and other factors within the TCL microenvironment activate JAK/STAT signaling, including IL-2, IL-6, and IL-15. As a result, JAK/STAT pathway activation within tumor cells is common. In fact, previous reports have described phosphorylated STAT3 (pSTAT3) within tumor cells in 27% of peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS); 29% of angioimmunoblastic T-cell lymphoma (AITL); and 57% of ALK− anaplastic large cell lymphoma (ALK− ALCL).11
In preclinical models, JAK/STAT mutations confer sensitivity to JAK inhibition. For example, treatment of STAT3 mutant ALCL cell lines with the JAK1/2 inhibition ruxolitinib led to abrogation of STAT3 phosphorylation and reduced cell proliferation.12 Furthermore, in mice bearing patient-derived xenografts generated from primary ALK− ALCL with JAK1 or STAT3 mutations, treatment with ruxolitinib caused significant inhibition of tumor growth.12 We previously showed similar effects upon treatment of mice engrafted with a T-PLL patient-derived xenograft that harbored a BCR-JAK2 fusion.13 Importantly, JAK inhibition caused dose-dependent inhibition of cell growth in STAT3- and STAT5B-mutated extranodal natural killer (NK)/T-cell lymphoma and γδ-TCL cell lines.10 This suggests that STAT3/5 mutations amplify upstream signals rather than drive JAK-independent growth.
Building upon the broad activation of JAK/STAT signaling in TCLs and preclinical data suggesting sensitivity to JAK inhibition, we aimed to investigate the efficacy of ruxolitinib in patients with relapsed and refractory TCL. We hypothesized that TCLs with JAK/STAT activation would be more likely to respond to ruxolitinib, and thus we designed the study to enrich for patients with tumors with genetic or phenotypic evidence of JAK/STAT activation.
Methods
Study design
This was a multicenter, investigator-initiated, phase 2 study that was carried out at Memorial Sloan Kettering Cancer Center, Dana-Farber Cancer Institute, University of Miami, and Weill Cornell Medical Center. Patients were enrolled onto 1 of 3 cohorts: Cohort 1: Patients whose tumors harbor known activating mutations in JAK1, JAK2, JAK3, STAT3, or STAT5B (allele frequency ≥0.1). Cohort 2: Patients whose tumors lack mutations in these factors but have functional evidence of JAK/STAT activation defined as ≥30% phospho-STAT3 staining in tumor cells by immunohistochemistry (IHC). Cohort 3: Patients who did not meet criteria for cohort 1 or 2 or who lacked adequate tissue for sequencing or assessment of pSTAT3 by IHC. Patients initially enrolled onto cohort 3 and determined to have activating JAK/STAT mutations or functional JAK/STAT activation after enrollment were moved to cohort 1 or 2 (Figure 1). The study was approved by the institutional review boards at all participating sites and registered at http://clinicaltrials.gov as NCT02974647.
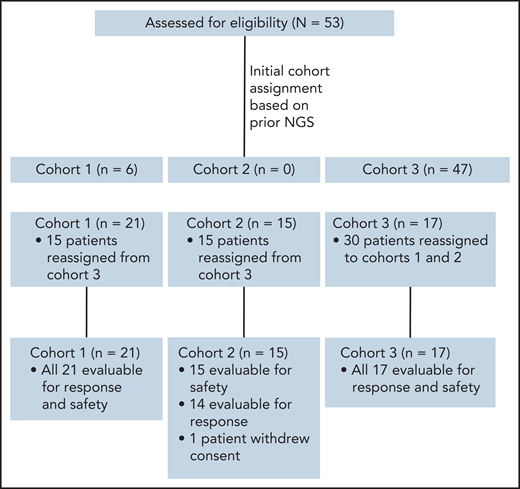
CONSORT diagram showing distribution of patients among cohorts upon initial enrollment, based on prior NGS performed on patient biopsies, and at final assignment after NGS of preenrollment biopsy and/or pSTAT3 IHC. Note that the final cohort for evaluation was 52 patients, as 1 patient in cohort 2 withdrew consent during the first week of treatment.
CONSORT diagram showing distribution of patients among cohorts upon initial enrollment, based on prior NGS performed on patient biopsies, and at final assignment after NGS of preenrollment biopsy and/or pSTAT3 IHC. Note that the final cohort for evaluation was 52 patients, as 1 patient in cohort 2 withdrew consent during the first week of treatment.
Patient eligibility
Patients age ≥18 years with peripheral T- or NK-cell lymphoma or cutaneous T-cell lymphoma (stage IB and greater) were eligible. Previously untreated patients with T-PLL were eligible (if age or comorbidities prohibited treatment with alemtuzumab); however, all other patients were required to have received ≥1 systemic therapy. Adequate organ function was required, defined as absolute neutrophil count of 1000/μL (500/μL for patients with neutropenia due to lymphoma); platelet count ≥100 × 109/L (≥50 × 109/L if due to lymphoma); hemoglobin ≥8 g/dL; bilirubin ≤1.5x upper limit of normal (ULN) or ≤3x ULN if due to lymphoma; AST and ALT ≤3x ULN (or ≤5x ULN if due to lymphoma); and creatinine clearance ≥30 mL/min (creatinine clearance of 15-29 mL/min allowed if baseline platelets ≥150 × 109/L).
Treatment
Patients received ruxolitinib 20 mg twice daily orally on 28-day cycles. Treatment continued until disease progression, unacceptable toxicity, or at the discretion of the treating physician. Dose reductions were recommended for grade ≥3 toxicities. Growth factor support and transfusions were allowed at the discretion of the investigator.
JAK/STAT pathway activation assessment
Baseline biopsies from each patient were subjected to next-generation sequencing (NGS) using the MSK integrated mutation profiling of actionable cancer targets platform or similar assays at participating institutions.14 Tumor samples were assessed by IHC for phosph-STAT3 (pSTAT3) (Clone M9C6; Cell Signaling, MA) by 1 hematopathologist (A.D). To define pSTAT3 activation, we used a cutoff of ≥30% based upon data from a series of TCL samples.11
Response assessment
Patients were assessed for response after cycle 2, cycle 5, and every 3 cycles thereafter. Methods used for response assessment were dependent upon subtype. Fluorodeoxyglucose-avid TCLs (such as PTCL, NOS; AITL; and ALCL) were assessed using Lugano Classification.15 MF was assessed using the Global Response Criteria.16 T-PLL was assessed using consensus criteria for T-PLL.17 T-LGL was assessed using a modification of the response assessment from the E5998 study,18 with complete response defined as normalization of blood counts (absolute neutrophil count >1500/μL, lymphocyte count <4000/μL, hemoglobin >11 g/dL, and platelet count >100 000/μL) along with normalization of T-LGL count (<400/μL) by flow cytometry. Partial response (PR) was defined as improvement in hematologic parameters in the absence of complete response (CR) (for patients initially neutropenic, absolute neutrophil count >500 as long as this represented 50% increase; for patients initially anemic, increase in hemoglobin by >1 g/dL for at least 4 months and decrease in monthly transfusion requirements of >50% for at least 4 months). Stable disease was defined as improvement in cytopenias (without meeting criteria for PR) for a minimum of 6 months.Progression of disease was defined as worsening hematologic parameters in patients previously achieving stable disease (SD), PR, or CR.
Multispectral immunofluorescence
Available pretreatment formalin-fixed paraffin-embedded tissue samples from patients enrolled in this clinical trial were analyzed with multiplex immunofluorescence (mIF) using the Vectra platform, and the data were extracted using HALO software (Indica Labs). Detailed methods for multiplex tissue staining and imaging are included in the supplemental Methods, available on the Blood Web site.
Statistical methods
The primary endpoint of the study was CBR, defined as a combination of CR, PR, and SD lasting at least 6 months (SD > 6 months). For sample size calculation and futility analysis, we used a slightly different endpoint: disease control rate, which is a combination of CR, PR, and SD lasting 8 weeks (2 cycles) of treatment. We chose this unconventional endpoint to adapt to the heterogeneity of diseases enrolled on this study that require different methods of response assessment and to avoid rejecting a drug with potential for clinical benefit in this patient population of high unmet need. A minimax Simon 2-stage design was used to determine the appropriate enrollment size for each cohort. For cohorts 1 and 2, we defined a disease control rate of 50% and an unacceptable disease control rate of 25% or lower. Accordingly, we initially enrolled 9 eligible patients to cohorts 1 and 2 in the first stage. If 7 or more patients had progression of disease within 2 cycles (first restaging), no additional patients would be enrolled, and the regimen would be considered not promising. If 6 or fewer patients had progressive disease, then an additional 8 patients would be enrolled to each cohort for the second stage. Among the total 17 patients, if 10 or fewer patients progressed within the first 8 weeks, then this treatment regimen would be declared promising. For cohort 3, we expected the response rate to be lower, and we defined the disease control rate to be 29% and an unacceptable disease control rate to be 10% or less. To this end, we initially enrolled 9 patients to cohort 3. If all patients had progressive disease within the first 2 cycles of treatment, this cohort would have been closed for futility. If 8 or fewer progressions were observed in the first 9 patients, an additional 9 patients would be enrolled. If 14 or fewer progressions were observed among the 18 patients, this treatment regimen would be declared promising. For all cohorts, this decision rule had a type I error rate of 0.10 and a type II error rate of 0.20. Enrollment to the study stopped once the overall accrual of evaluable patients reached 52, which was the sum of patients needed across all 3 cohorts.
The protocol allowed for moving of patients from 1 cohort to another as genomic evidence emerged during the trial. Design rules were recalculated for cohort 2 based on the realized sample size of 14, and the original type I and type II error probabilities, which resulted in a decision rule of 8 or fewer progressors signaling a promising result.
Secondary endpoints included toxicity graded according to the Common Terminology Criteria for Adverse Events (version 4.0), progression-free survival (PFS), overall survival (OS), and duration of response. PFS and OS were measured from the time of initiation of treatment by the Kaplan-Meier method. Correlation between assigned cohort and response was assessed using the 2-sided Fisher’s exact test. Differences in mIF marker and cytokine expression by response status were assessed with the Wilcoxon test. Receiver operating characteristic curve was performed for tumor cell expression of pS6 and overall response rate (ORR) to determine the optimal cutoff for defining positivity. Statistical analysis was done using R version 4.3.
All patients provided informed consent per the Declaration of Helsinki.
Results
Overall patient characteristics
Between January 2017 and July 2019, 53 patients enrolled across 4 institutions, including 21 in cohort 1, 15 in cohort 2, and 17 in cohort 3. Patient characteristics according to cohort are detailed in Table 1; genetic sequencing, IHC, and mIF results are provided in supplemental Table 1. In summary, 51% of patients were males, median age was 62 (19-88), and median number of prior therapies was 3 (range 0-11). The most common disease subtypes included PTCL, NOS (n = 12); T-PLL (n = 8); AITL/TFH (n = 9); and T-LGL (n = 5).
Patient characteristics
| Characteristics | Total (n = 53) | Cohort 1 (n = 21) | Cohort 2 (n = 15) | Cohort 3 (n = 17) | P |
|---|---|---|---|---|---|
| Median age (range) | 62 (19-88) | 63 (47-88) | 69 (44-78) | 57 (19-76) | .042 |
| Male, n (%) | 27 (51%) | 11 (52%) | 7 (47%) | 9 (53%) | .9 |
| Prior no. of therapies, median (range) | 3 (0-11) | 2 (0-9) | 4 (1-11) | 4 (1-8) | <.001 |
| Prior transplants, n (%) | 12 (23%) | 3 (14%) | 3 (20%) | 6 (35%) | .3 |
| Autologous | 6 (11%) | 1 (5%) | 2 (13%) | 3 (18%) | .4 |
| Allogeneic | 6 (11%) | 2 (10%) | 1 (7%) | 3 (18%) | .6 |
| Race, n (%) | .5 | ||||
| White | 42 (79%) | 16 (76%) | 13 (87%) | 13 (76%) | |
| African American or Black | 5 (9%) | 1 (5%) | 1 (7%) | 3 (18%) | |
| Asian | 3 (6%) | 3 (14%) | 0 | 0 | |
| Unknown | 3 (6%) | 1 (5%) | 1 (7%) | 1 (6%) |
| Characteristics | Total (n = 53) | Cohort 1 (n = 21) | Cohort 2 (n = 15) | Cohort 3 (n = 17) | P |
|---|---|---|---|---|---|
| Median age (range) | 62 (19-88) | 63 (47-88) | 69 (44-78) | 57 (19-76) | .042 |
| Male, n (%) | 27 (51%) | 11 (52%) | 7 (47%) | 9 (53%) | .9 |
| Prior no. of therapies, median (range) | 3 (0-11) | 2 (0-9) | 4 (1-11) | 4 (1-8) | <.001 |
| Prior transplants, n (%) | 12 (23%) | 3 (14%) | 3 (20%) | 6 (35%) | .3 |
| Autologous | 6 (11%) | 1 (5%) | 2 (13%) | 3 (18%) | .4 |
| Allogeneic | 6 (11%) | 2 (10%) | 1 (7%) | 3 (18%) | .6 |
| Race, n (%) | .5 | ||||
| White | 42 (79%) | 16 (76%) | 13 (87%) | 13 (76%) | |
| African American or Black | 5 (9%) | 1 (5%) | 1 (7%) | 3 (18%) | |
| Asian | 3 (6%) | 3 (14%) | 0 | 0 | |
| Unknown | 3 (6%) | 1 (5%) | 1 (7%) | 1 (6%) |
| Subtype, n (%) | Total (n=53) | Cohort 1 (n=21) | Cohort 2 (n=15) | Cohort 3 (n=17) | P |
|---|---|---|---|---|---|
| PTCL, NOS | 12 (23%) | 2 (10%) | 5 (33%) | 5 (29%) | .2 |
| T-PLL | 8 (15%) | 7 (33%) | 1 (7%) | 0 | .007 |
| AITL/TFH | 9 (17%) | 2 (10%) | 5 (33%) | 2 (12%) | .2 |
| T-LGL | 5 (9%) | 3 (14%) | 0 | 2 (12%) | .4 |
| ALCL* | 4 (8%) | 2 (10%) | 0 | 2 (12%) | .5 |
| ATLL | 3 (6%) | 0 | 0 | 3 (18%) | .049 |
| MF† | 7 (13%) | 2 (10%) | 3 (20%) | 2 (12%) | .7 |
| γ/δ TCLs‡ | 4 (8%) | 3 (14%) | 0 (0%) | 1 (6%) | .4 |
| SPTCL | 1 (2%) | 0 | 1 (7%) | 0 | .3 |
| Subtype, n (%) | Total (n=53) | Cohort 1 (n=21) | Cohort 2 (n=15) | Cohort 3 (n=17) | P |
|---|---|---|---|---|---|
| PTCL, NOS | 12 (23%) | 2 (10%) | 5 (33%) | 5 (29%) | .2 |
| T-PLL | 8 (15%) | 7 (33%) | 1 (7%) | 0 | .007 |
| AITL/TFH | 9 (17%) | 2 (10%) | 5 (33%) | 2 (12%) | .2 |
| T-LGL | 5 (9%) | 3 (14%) | 0 | 2 (12%) | .4 |
| ALCL* | 4 (8%) | 2 (10%) | 0 | 2 (12%) | .5 |
| ATLL | 3 (6%) | 0 | 0 | 3 (18%) | .049 |
| MF† | 7 (13%) | 2 (10%) | 3 (20%) | 2 (12%) | .7 |
| γ/δ TCLs‡ | 4 (8%) | 3 (14%) | 0 (0%) | 1 (6%) | .4 |
| SPTCL | 1 (2%) | 0 | 1 (7%) | 0 | .3 |
AITL/TFH, angioimmunoblastic T-cell lymphoma and other T-follicular helper lymphomas; ATLL, adult T-cell lymphoma lymphoma/leukemia; γ/δ TCL, γ/δ T-cell lymphomas; SPTCL, subcutaneous panniculitis-like T-cell lymphoma.
ALCL includes systemic ALK− ALCL (n = 3) and primary cutaneous ALCL (n = 1).
2 patients with MF had large cell transformation.
γ/δ TCLs included hepatosplenic T-cell lymphoma (n = 2), monomorphic epitheoliotropic intestinal T-cell lymphoma (n = 1), and primary cutaneous γ/δ T-cell lymphoma (n = 1).
Cohort 1 characteristics
Among patients in cohort 1, mutated genes included STAT5B (n = 8), STAT3 (n = 7), JAK1 (n = 2), JAK2 (n = 1), and JAK3 (n = 6) (Figure 2). Of these patients, 2 were positive for pSTAT3 in ≥30% of tumor cells by IHC (1 with a STAT3 mutation and 1 with both a STAT3 and JAK1 mutation), 12 were negative for pSTAT3 expression, and 7 did not have IHC for pSTAT3 performed due to lack of adequate tissue availability. Among the 12 who were negative for pSTAT3, median pSTAT3 expression was 5% of tumor cells (range 0% to 20%).
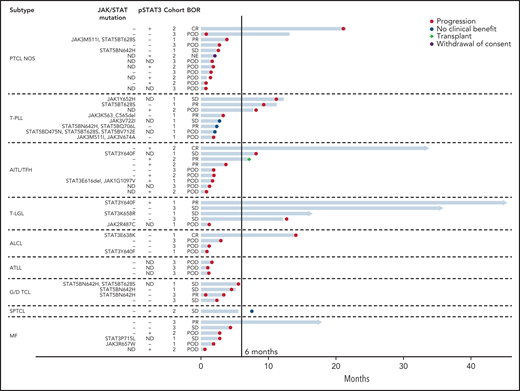
Swimmer’s plot for all evaluable patients treated with ruxolitinib. Each bar represents 1 subject in the study. Right arrow cap indicates ongoing treatment. ALCL included systemic ALK− ALCL (n = 3) and primary cutaneous ALCL (n = 1). G/D TCL included hepatosplenic T-cell lymphoma (n = 2), monomorphic epitheliotropic intestinal T-cell lymphoma (n = 1), and primary cutaneous γδ-TCL (n = 1). JAK/STAT Mutations: only JAK or STAT mutations listed. (−) indicates no JAK or STAT mutation present. Additional mutations identified by next generation sequencing (NGS) are provided in supplemental Table 1. pSTAT3: (+) is defined as ≥30% expression in tumor cells by immunohistochemical staining; (−) defined as <30% expression. ND indicates that NGS or IHC for pSTAT3 was not done. Cohorts: cohort 1 (JAK/STAT mutations present); cohort 2 (pSTAT3 ≥30% by IHC); cohort 3 (neither or ND). AITL/TFH, angioimmunoblastic T-cell lymphoma and other T-follicular helper lymphomas; ALK− ALCL, ALK− anaplastic large cell lymphoma; BOR, best overall response; POD, progression of disease.
Swimmer’s plot for all evaluable patients treated with ruxolitinib. Each bar represents 1 subject in the study. Right arrow cap indicates ongoing treatment. ALCL included systemic ALK− ALCL (n = 3) and primary cutaneous ALCL (n = 1). G/D TCL included hepatosplenic T-cell lymphoma (n = 2), monomorphic epitheliotropic intestinal T-cell lymphoma (n = 1), and primary cutaneous γδ-TCL (n = 1). JAK/STAT Mutations: only JAK or STAT mutations listed. (−) indicates no JAK or STAT mutation present. Additional mutations identified by next generation sequencing (NGS) are provided in supplemental Table 1. pSTAT3: (+) is defined as ≥30% expression in tumor cells by immunohistochemical staining; (−) defined as <30% expression. ND indicates that NGS or IHC for pSTAT3 was not done. Cohorts: cohort 1 (JAK/STAT mutations present); cohort 2 (pSTAT3 ≥30% by IHC); cohort 3 (neither or ND). AITL/TFH, angioimmunoblastic T-cell lymphoma and other T-follicular helper lymphomas; ALK− ALCL, ALK− anaplastic large cell lymphoma; BOR, best overall response; POD, progression of disease.
Cohort 2 characteristics
In cohort 2, the median percent of tumor cells that were pSTAT3+ by IHC was 65% (range 30% to 100%) (supplemental Table 1). NGS was completed on biopsies from 9 of 15 patients in cohort 2 and showed no JAK or STAT mutations. The other 6 patients did not have NGS done due to inadequate tissue (Figure 2).
Cohort 3 characteristics
Six of 17 patients in cohort 3 had incomplete JAK/STAT activation data, including 3 patients with NGS but missing IHC for pSTAT3 and 3 patients missing both NGS and IHC (Figure 2).
Overall efficacy
Among the 53 patients, 52 were evaluable for response (1 from cohort 2 withdrew consent following only 1 week of treatment without progression of disease or toxicity) (Table 2; Figure 2). Overall, there were 3 (6%) CRs, 10 (19%) PRs, and 5 (10%) with stable disease lasting at least 6 months (SD >6 months). The ORR and CBR were 25% and 35%, respectively. CBRs in cohorts 1, 2, and 3 were 48%, 36%, and 18%, respectively (cohorts 1 & 2 vs 3, P = .073). ORR in cohorts 1, 2, and 3 were 33%, 29%, and 12%, respectively (cohorts 1 & 2 vs 3, P = .2). For the 45 patients with PTCL (ie, excluding the 7 patients with mycosis fungoides), CBRs in cohorts 1, 2, and 3 were 53%, 45%, and 13%, respectively (cohorts 1 & 2 vs 3, P = .02) (Table 3).
Efficacy according to cohort and subtype
| Cohorts | Total treated | Total evaluable for response | ORR | CBR | CR | PR | SD >6 mo |
|---|---|---|---|---|---|---|---|
| Cohort 1 | 21 | 21 | 7 (33%) | 10 (48%) | 1 (5%) | 6 (29%) | 3 (14%) |
| Cohort 2 | 15 | 14 | 4 (29%) | 5 (36%) | 2 (14%) | 2 (14%) | 1 (7%) |
| Cohort 3 | 17 | 17 | 2 (12%) | 3 (18%) | 0 | 2 (12%) | 1 (6%) |
| Total | 53 | 52 | 13 (25%) | 18 (35%) | 3 (6%) | 10 (19%) | 5 (10%) |
| P (cohorts 1 & 2 vs 3) | P = .2 | P = .073 | |||||
| Cohorts | Total treated | Total evaluable for response | ORR | CBR | CR | PR | SD >6 mo |
|---|---|---|---|---|---|---|---|
| Cohort 1 | 21 | 21 | 7 (33%) | 10 (48%) | 1 (5%) | 6 (29%) | 3 (14%) |
| Cohort 2 | 15 | 14 | 4 (29%) | 5 (36%) | 2 (14%) | 2 (14%) | 1 (7%) |
| Cohort 3 | 17 | 17 | 2 (12%) | 3 (18%) | 0 | 2 (12%) | 1 (6%) |
| Total | 53 | 52 | 13 (25%) | 18 (35%) | 3 (6%) | 10 (19%) | 5 (10%) |
| P (cohorts 1 & 2 vs 3) | P = .2 | P = .073 | |||||
| Subtypes | Total treated | Total evaluable for response | ORR | CBR | CR | PR | SD >6 mo |
|---|---|---|---|---|---|---|---|
| PTCL, NOS | 12 | 11 | 2 (18%) | 2 (18%) | 1 (9%) | 1 (9%) | 0 |
| T-PLL | 8 | 8 | 3 (38%) | 4 (50%) | 0 | 3 (38%) | 1 (13%) |
| AITL/TFH | 9 | 9 | 3 (33%) | 4 (44%) | 1 (11%) | 2 (22%) | 1 (11%) |
| T-LGL | 5 | 5 | 2 (40%) | 4 (80%) | 0 | 2 (40%) | 2 (40%) |
| ALCL | 4 | 4 | 1 (25%) | 1 (25%) | 1 (25%) | 0 | 0 |
| ATLL | 3 | 3 | 0 | 0 | 0 | 0 | 0 |
| MF | 7 | 7 | 1 (14%) | 1 (14%) | 0 | 1 (14%) | 0 |
| γ/δ TCLs | 4 | 4 | 1 (25%) | 1 (25%) | 0 | 1 (25%) | 0 (0%) |
| SPTCL | 1 | 1 | 0 | 1 (100%) | 0 | 0 | 1(100%) |
| Subtypes | Total treated | Total evaluable for response | ORR | CBR | CR | PR | SD >6 mo |
|---|---|---|---|---|---|---|---|
| PTCL, NOS | 12 | 11 | 2 (18%) | 2 (18%) | 1 (9%) | 1 (9%) | 0 |
| T-PLL | 8 | 8 | 3 (38%) | 4 (50%) | 0 | 3 (38%) | 1 (13%) |
| AITL/TFH | 9 | 9 | 3 (33%) | 4 (44%) | 1 (11%) | 2 (22%) | 1 (11%) |
| T-LGL | 5 | 5 | 2 (40%) | 4 (80%) | 0 | 2 (40%) | 2 (40%) |
| ALCL | 4 | 4 | 1 (25%) | 1 (25%) | 1 (25%) | 0 | 0 |
| ATLL | 3 | 3 | 0 | 0 | 0 | 0 | 0 |
| MF | 7 | 7 | 1 (14%) | 1 (14%) | 0 | 1 (14%) | 0 |
| γ/δ TCLs | 4 | 4 | 1 (25%) | 1 (25%) | 0 | 1 (25%) | 0 (0%) |
| SPTCL | 1 | 1 | 0 | 1 (100%) | 0 | 0 | 1(100%) |
AITL/TFH, angioimmunoblastic T-cell lymphoma and other T-follicular helper lymphomas; ATLL, adult T-cell lymphoma lymphoma/leukemia; γ/δ TCL, γ/δ T-cell lymphomas; SPTCL, subcutaneous panniculitis-like T-cell lymphoma.
Efficacy for PTCL according to cohort
| Cohorts | Total treated | Total evaluable for response | ORR | CBR | CR | PR | SD >6 mo |
|---|---|---|---|---|---|---|---|
| Cohort 1 | 19 | 19 | 7 (37%) | 10 (53%) | 1 (5%) | 6 (32%) | 3 (16%) |
| Cohort 2 | 12 | 11 | 4 (36%) | 5 (45%) | 2 (18%) | 2 (18%) | 1 (9%) |
| Cohort 3 | 15 | 15 | 1 (7%) | 2 (13%) | 0 (0%) | 1 (7%) | 1 (7%) |
| Total | 46 | 45 | 12 (27%) | 17 (38%) | 3 (7%) | 9 (20%) | 5 (11%) |
| P (cohorts 1 & 2 vs 3) | ORR, P = .04 | CBR, P = .02 | |||||
| Cohorts | Total treated | Total evaluable for response | ORR | CBR | CR | PR | SD >6 mo |
|---|---|---|---|---|---|---|---|
| Cohort 1 | 19 | 19 | 7 (37%) | 10 (53%) | 1 (5%) | 6 (32%) | 3 (16%) |
| Cohort 2 | 12 | 11 | 4 (36%) | 5 (45%) | 2 (18%) | 2 (18%) | 1 (9%) |
| Cohort 3 | 15 | 15 | 1 (7%) | 2 (13%) | 0 (0%) | 1 (7%) | 1 (7%) |
| Total | 46 | 45 | 12 (27%) | 17 (38%) | 3 (7%) | 9 (20%) | 5 (11%) |
| P (cohorts 1 & 2 vs 3) | ORR, P = .04 | CBR, P = .02 | |||||
Median time to best response was 6.3 months (range: 5.5-7.2 months). Median PFS and OS were 2.8 months (95% CI: 1.8-4.5 months) and 26.2 months (95% CI: 11.5-not reached [NR]), respectively. Median duration of response was 8.4 months (95% CI: 7.5-NR). Median duration of response by cohort were 7.5, 14.7 months, and NR for cohorts 1, 2, and 3, respectively (P = .3). Eight patients achieved responses lasting >1 year and 5 remain on therapy (Figure 2). These 8 exceptional responders included 4 of 5 with T-LGL (progression-free 12.65-45.4+ months); 1 with PTCL, NOS (progression-free 21 months); 1 with AITL (progression-free 33.82+ months); 1 with primary cutaneous ALCL (progression-free 14.0 months); and 1 with MF (progression-free 17.95+ months).
Efficacy according to subtype
The most common PTCL entities; PTCL, NOS; AITL; and ALCL, represented 24 (45%) of all patients enrolled on the study. Among these patients, 23 were evaluable for response and 5, 9, and 9 were in cohorts 1, 2, and 3, respectively. CBR was significantly higher among patients in cohorts 1 and 2 compared with cohort 3 (P = .048) (supplemental Table 2).
Among the 5 patients with T-LGL, CBR and ORR were 80% and 40%, respectively. PFS was >1 year for all 4 responders. Two of the patients with T-LGL had STAT3 mutations (both responded to ruxolitinib) while a third patient had a JAK2 mutation (no response to ruxolitinib). The other 2 patients with T-LGL lacked mutations or pSTAT3 expression by IHC and both responded to ruxolitinib (Figure 2).
All 8 patients with T-PLL had evidence of JAK/STAT activation, including 7 patients with JAK or STAT mutations and 1 with only pSTAT3 expression. The ORR and CBR among patients with T-PLL were 38% and 50%, respectively.
Among the 7 patients with MF, 2 had JAK or STAT mutations, 3 had pSTAT3 expression (and no JAK/STAT mutations), and 2 had neither (Figure 2). There was only 1 responder (with ongoing PR for 17.95+ months),a patient whose tumor had borderline pSTAT overexpression (20%) and lacked JAK/STAT mutations.
Safety
All 53 patients were evaluable for safety. Adverse events affecting >5% of patients are shown in Table 4. Adverse events were consistent with the known side effect profile of ruxolitinib and primarily involved cytopenias. Treatment-related serious adverse events included herpes simplex virus-1 stomatitis (n = 1), spontaneous bacterial peritonitis (n = 1), febrile neutropenia (n = 3), anemia (n = 1), and herpes zoster (n = 1).
Adverse events occurring in less than 5% of patients
| Adverse events | No. of patients | ||||
|---|---|---|---|---|---|
| Possible, probably or definitely related toxicities | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total (%) |
| Febrile neutropenia | 1 | 2 | 3 (6%) | ||
| Fatigue | 3 | 2 | 5 (9%) | ||
| Diarrhea | 5 | 2 | 7 (13%) | ||
| Platelet count decreased | 3 | 1 | 4 | 1 | 9 (17%) |
| Neutrophil count decreased | 5 | 5 | 10 (19%) | ||
| Anemia | 3 | 3 | 9 | 15 (28%) | |
| Adverse events | No. of patients | ||||
|---|---|---|---|---|---|
| Possible, probably or definitely related toxicities | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total (%) |
| Febrile neutropenia | 1 | 2 | 3 (6%) | ||
| Fatigue | 3 | 2 | 5 (9%) | ||
| Diarrhea | 5 | 2 | 7 (13%) | ||
| Platelet count decreased | 3 | 1 | 4 | 1 | 9 (17%) |
| Neutrophil count decreased | 5 | 5 | 10 (19%) | ||
| Anemia | 3 | 3 | 9 | 15 (28%) | |
Multispectral immunofluorescence
Pretreatment tissue was available for mIF staining from 9 patients (3 with CBR and 6 with no CBR). Figure 3A shows a representative image from a patient with MF demonstrating spatial definition of the area of tumor involvement for assessment. Figure 3B shows staining of a PTCL sample with 6 antibodies plus DAPI. The median number of total TCL cells in pretreatment biopsies was 960 (range, 97-3889), and the median TCL cell fraction within involved areas was 28.19% (range, 2.13% to 91.35%). The median macrophage fraction within involved areas was 3.59% (range 1.06% to 23.35%).
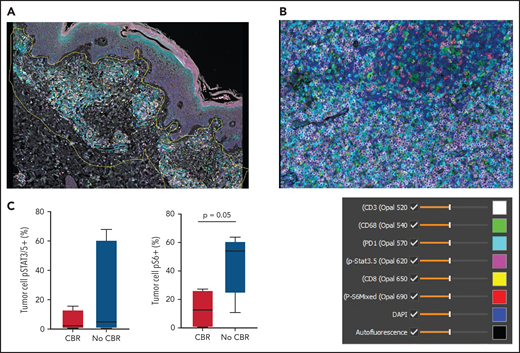
Multispectral immunofluorescence with 6 markers plus 4′,6-diamidino-2-phenylindole (DAPI) using the Vectra platform in pretreatment biopsies from 9 patients. (A) Spatial definition of the tumor involvement in a biopsy of MF using the HALO software. (B) Representative mIF image from a biopsy of PTCL stained as indicated. (C) Phospho-S6 expression in tumor cells distinguished patients with CBR or no CBR from ruxolitinib (P = .05). Phospho-STAT3/5 expression in tumor cells did not differ between patients with CBR or no CBR (P = .9).
Multispectral immunofluorescence with 6 markers plus 4′,6-diamidino-2-phenylindole (DAPI) using the Vectra platform in pretreatment biopsies from 9 patients. (A) Spatial definition of the tumor involvement in a biopsy of MF using the HALO software. (B) Representative mIF image from a biopsy of PTCL stained as indicated. (C) Phospho-S6 expression in tumor cells distinguished patients with CBR or no CBR from ruxolitinib (P = .05). Phospho-STAT3/5 expression in tumor cells did not differ between patients with CBR or no CBR (P = .9).
Discussion
Given the significant unmet need for new therapies to treat relapsed/refractory TCLs and the prevalence of JAK/STAT pathway activation across numerous TCL entities, a phase 2 study of ruxolitinib in TCL was a logical pursuit. This study showed broad activity across various TCL subtypes, including 8 (15%) patients with responses lasting more than 1 year, and provides proof of principle that the JAK/STAT pathway is a worthwhile target in TCL.
We hypothesized that JAK/STAT pathway activation would predict for sensitivity to ruxolitinib, and therefore we designed the study to enrich for entities with JAK or STAT mutations as well as IHC evidence of JAK/STAT pathway activation. We enrolled patients onto 3 cohorts defined by presence of JAK/STAT mutations (cohort 1), pSTAT3 expression in ≥30% of tumor cells (cohort 2), or neither (cohort 3). Each cohort included patients with markedly heterogeneous varieties of TCL, which made it difficult to analyze patients within each cohort together. In fact, based upon the predefined decision rules for each cohort, clinical efficacy was promising in cohorts 1 and 3 but not in cohort 2 (due to 9 progressors within cohort 2, which was over the recalculated design threshold of 8). Nevertheless, among PTCL and non-MF CTCL, the presence of JAK/STAT mutations and/or pSTAT3 expression in ≥30% of tumor cells predicted for CBR (P = .02). In contrast, among patients with MF, despite 5/7 (71%) showing evidence of JAK/STAT activation, only 1 patient (whose tumor showed pSTAT3 expression in only 20% of tumor cells) responded. These observations suggest that JAK/STAT activation can be used as a biomarker of response to ruxolitinib in PTCL and non-MF CTCL. In MF as well as ATLL (0 of 3 had CBR), despite high rates of JAK/STAT activation, ruxolitinib is unlikely to be effective as a single agent.
In 2 entities commonly associated with JAK/STAT activation, T-PLL and T-LGL, CBRs were 50% and 80%, respectively. T-PLL is a rare, aggressive disease with limited treatment options and frequent JAK or STAT mutations. The efficacy we observed in T-PLL is encouraging; however, durability was limited, and ruxolitinib-based combinations may turn out to be more promising. Studies in primary T-PLL cells show that JAK inhibition can enhance BCL-2–dependent survival, suggesting that venetoclax may represent a promising combination partner for ruxolitinib in this disease.19 With regard to T-LGL, despite frequently being associated with STAT3 mutations, responses were observed regardless of JAK/STAT genotype. This suggests that certain entities, such as T-LGL, may be intrinsically sensitive to JAK inhibition regardless of the presence or absence of JAK/STAT activating mutations. An important limitation of this study, though, was our inability to assess pSTAT3 expression in the peripheral blood of patients with T-LGL and T-PLL. Instead, pSTAT3 expression was assessed on bone marrow, which may be less accurate due to harsh conditions from decalcification.
A striking finding was the lack of pSTAT3 staining by IHC in 12 of 14 evaluable cases with JAK/STAT mutations. At the same time, 9 of 11 cases with pSTAT3 staining (and NGS data) lacked JAK/STAT mutations. This suggests that mutations may not markedly upregulate pSTAT3 within tumor biopsies. Instead, nontumor cell autonomous signals from the microenvironment may have greater effects. In either case, it may be necessary to perform both sequencing and IHC to fully identify which patients could benefit from ruxolitinib.
While the lack of JAK/STAT activation appears to predict for reduced sensitivity to ruxolitinib in PTCL, the presence of activation was not a guarantee of response. One potential reason could be due to the limitations of ruxolitinib, which causes thrombocytopenia and anemia through inhibition of JAK2, thus limiting dose escalation. In fact, preliminary results from a phase 1/2 study evaluating a JAK1 inhibitor (DZD4205) in patients with primarily PTCL, NOS and AITL appears promising (ORR 41%) and possibly superior to this present study; however, it is not possible to compare, as the study populations differ and data regarding JAK/STAT mutation status was not provided.20 Ruxolitinib has nearly equipotent activity against JAK1 and JAK2 but lacks potency against JAK3. Thus, it is possible that a broader JAK inhibitor could further suppress cytokine signaling. Arguing against this, most of the essential cytokines in TCLs involve common γ-chain signaling (eg, IL-2, IL-15, and IL-21), which requires JAK1.21 Alternative strategies for targeting the pathway, such as with STAT3-directed, heterobifunctional degraders, may also have promise.22,23
It is likely that in some cases, despite genetic or IHC evidence of JAK/STAT activation, other oncogenic pathways may be in play that potentially drive resistance to JAK inhibition. One potential example is the PI3K pathway, which was recently shown to be clinically relevant in TCL. A phase 1 study of the oral PI3K-δ/γ inhibitor duvelisib showed ORRs of 50% and 32% in systemic TCL (n = 16) and cutaneous T-cell lymphoma (n = 19), respectively, demonstrating the importance of the PI3K pathway as an oncogenic driver in these diseases.24 The T-cell receptor (TCR) pathway is a target of frequent mutations in TCLs. In contrast with cytokine receptors that activate JAK/STAT, TCR signaling activates MAP kinase and PI3K signaling.25 During T-cell differentiation, the effects of JAK/STAT signaling (eg, downstream of the IL2 receptor) are integrated with signaling through the TCR. However, strong activation of both JAK/STAT and MAPK/PI3K has been shown to counteract malignant transformation of lymphocytes, suggesting that activation of the 2 pathways is likely mutually exclusive in individual tumor cells.26 The extent to which activation of MAPK/PI3K signaling predicts against JAK/STAT activation in TCLs has not been explored. Interestingly, preclinical data in TCL cell lines demonstrated cooperativity between PI3K and JAK/STAT signaling in maintaining downstream cell survival and proliferation signals, indicating that in some cases, targeting both pathways may be essential.27
A potential limitation of our study is how we chose to define JAK/STAT activation. Additional factors that can affect JAK/STAT activation (eg, mutations involving cytokine receptors and negative regulators) were not included in our cohort assignments. For IHC, we used only pSTAT3, as pSTAT5 staining by IHC was unreliable. In assessing expression by IHC, there is potential for interobserver disagreement, and it is also possible that 30% is not the optimal cutoff for pSTAT3 expression. In fact, one of the exceptional responders in this study was a patient with MF whose tumor showed 20% pSTAT3 expression by IHC, indicating that perhaps a lower cutoff may be appropriate for predicting response to ruxolitinib. Studies incorporating automated methods for estimating IHC staining in large series are needed to more accurately define the optimal cutoff. As mentioned above, the accuracy of pSTAT3 staining may vary between lymph nodes and bone marrow specimens, thus limiting our ability to confirm pSTAT3 expression for the leukemic diseases T-PLL and T-LGL, since only bone marrow was available for these patients. This would clearly impact our ability to evaluate the association between JAK/STAT activation and ruxolitinib efficacy in T-LGL. Newer methods such as phospho-flow may eventually allow us to more accurately assess pSTAT3 expression in fresh biopsies. The same concern also affects our exploratory analysis using mIF, which showed that baseline expression of pS6 predicted for lack of response to ruxolitinib. Much larger cohorts will be needed to determine whether pS6 can be used as a predictive biomarker and whether 25% is the most appropriate cutoff.
A significant limitation of our study was the limited tissue available for genetic, IHC, and mIF assessment. Biopsies of TCL are commonly performed using core needles, which provide limited tissue for clinical assessment and research correlative assays. In settings where predictive biomarkers could guide therapeutic selection, a stronger argument can be made for larger biopsies (when safe). Nonetheless, this study provides proof of principle that the JAK/STAT pathway is clinically relevant in TCLs. We demonstrate that the readily available oral inhibitor ruxolitinib was active across various subtypes characterized by JAK/STAT pathway activity with acceptable toxicity. Furthermore, in T-LGL, ruxolitinib provided benefit regardless of JAK/STAT mutational status. Based upon the small number of patients enrolled with T-LGL, ruxolitinib appears promising as a single agent, and further evaluation is warranted. For most other entities, further investigation is needed to identify which patients are likely to have exceptional responses to single-agent ruxolitinib. At the same time, the association between pS6 expression and response to ruxolitinib suggests that active PI3K/mTOR signaling confers intrinsic resistance to ruxolitinib and provides rationale for combination therapy targeting JAK and PI3K. Our upcoming study with ruxolitinib plus duvelisib in TCLs will test this hypothesis.
Acknowledgments
The authors gratefully acknowledge the members of the Molecular Diagnostics Service in the MSKCC Department of Pathology.
A.J.M. is a Scholar in Clinical Research of The Leukemia & Lymphoma Society.
These studies were supported by Specialized Center for Research grants from the Leukemia and Lymphoma Society (7011-16 and 7026-21 [D.M.W.]); a Career Development Award from the Leukemia and Lymphoma Society (2332-20 [A.J.M.]); grants from the National Institutes of Health/National Cancer Institute (NIH/NCI; P01 CA248384 and R35 CA2319858 [D.M.W.]), NIH/NCI MSK Lymphoma SPORE (P50 CA192937 [A.D.]); and the Farmer Family Foundation (A.D.). This study is an investigator-initiated study funded in part by Incyte.
Authorship
Contribution: A.J.M., S.M.H., D.M.W., and S.V. designed research; A.J.M., E.J., J. Ruan, J.H.S., S.N., P.M., S.V., N.G., H.H., T.D., L.P., S.R., A.S., N.Y., S.S., N.G., R.N., J.D., J.B., B.L., W.B., O.O., L.P., M.B.G., A.N., D.S., P.K., A.D., T.H., J. Rademaker, H.S., G.I., D.M.W., S.M.H. performed the research; A.J.M., N.G., E.D., S.M.H., D.M.W., and P.G. analyzed the data; and A.J.M., S.M.H., and D.M.W. wrote the paper.
Conflict-of-interest disclosure: A.J.M. reports research support from ADC Therapeutics, Beigene, Miragen, Seattle Genetics, Merck, Bristol-Myers Squibb, and Incyte; and consulting fees from Imbrium Therapeutics L.P./Purdue, Janpix Ltd., Merck, Seattle Genetics, and Takeda. D.M.W. reports research support from Daiichi Sankyo, Verastem, and Abcuro; consulting fees from AstraZeneca, Daiichi Sankyo, and Vor; advisory board membership for Travera, Trillium, Bantam, Ajax, and Secura; and is a founder of Travera and Ajax. A.D. reports personal fees from Physicians' Education Resource, Seattle Genetics, Takeda, Roche, EUSA Pharma, and Peer View; and research support from Roche and Takeda. M.B.G. reports research support from Amgen and Actinium Pharmaceuticals and advisory board participation for Sanofi. S.M.H. reports research support from ADC Therapeutics, Affimed, Aileron, Celgene, Crispr Therapeutics, Daiichi Sankyo, Forty Seven, Inc., Kyowa Hakko Kirin, Millennium/Takeda, Seattle Genetics, Trillium Therapeutics, and Verastem/SecuraBio; and consulting fees from Acrotech Biopharma, ADC Therapeutics, Astex, C4 Therapeutics, Celgene, Janssen, Kura Oncology, Kyowa Hakko Kirin, Myeloid Therapeutics, ONO Pharmaceuticals, Seattle Genetics, SecuraBio, Shoreline Biosciences, Inc., Takeda, Trillium Therapeutics, Tubulis, Verastem, and Vividion Therapeutics. S.V. is on the advisory board for Immunai and has received consulting fees from ADC Therapeutics. J. Ruan reports research support from BMS, AstraZeneca, Daiichi Sankyo, Pharmacyclics, and Seagen; and consultancy fees from BMS, Kite Pharma, Daiichi Sankyo, and Seagen. S.N. has participated in a medical advisory board for Kyowa Kirin. T.H. receives research support from the Parker Institute for Cancer Immunotherapy, Bristol Myers Squibb, and Calico Labs. The remaining authors declare no competing financial interests.
Correspondence: Alison Moskowitz, Memorial Sloan-Kettering Cancer Center, 530 East 74th St, New York, NY 10021; e-mail: moskowia@mskcc.org.
Presented in abstract form at the 2018 and 2019 annual meetings of the American Society of Hematology.
Deidentified individual participant data reported in the manuscript will be shared under the terms of a Data Use Agreement and may only be used for approved proposals. Requests may be made to: crdatashare@mskcc.org.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal