

Key Points
Engineered neutrophil-primed progenitors established from human iPSCs can produce neutrophil-like cells on a clinically applicable scale.
iPSC-derived neutrophil-like cells recruit immune cells and can prevent lethal infections.

Abstract
Neutrophils play an essential role in innate immune responses to bacterial and fungal infections, and loss of neutrophil function can increase the risk of acquiring lethal infections in clinical settings. Here, we show that engineered neutrophil-primed progenitors derived from human induced pluripotent stem cells can produce functional neutrophil-like cells at a clinically applicable scale that can act rapidly in vivo against lethal bacterial infections. Using 5 different mouse models, we systematically demonstrated that these neutrophil-like cells migrate to sites of inflammation and infection and increase survival against bacterial infection. In addition, we found that these human neutrophil-like cells can recruit murine immune cells. This system potentially provides a straight-forward solution for patients with neutrophil deficiency: an off-the-shelf neutrophil transfusion. This platform should facilitate the administration of human neutrophils for a broad spectrum of physiological and pathological conditions.
Introduction
Neutrophils are the most abundant leukocytes in human blood, performing several regulatory functions under a variety of physiological and pathological conditions,1 including infection,2 injury,3 autoimmunity,4 and cancer.5 Neutrophils bidirectionally crosstalk with other immune cells and amplify responses to infections in both innate and adaptive immunity. They act as first-line defenders against bacteria and fungi and provide support for immune responses to parasites and viruses.2,6 Dysregulation of neutrophils can critically affect the defense against infections and may result in death. Despite advances in supportive therapies, including antimicrobial drugs and neutrophil-stimulating agents, bacterial and fungal infections pose threats to patients with neutrophil dysregulation.7-10
Here, we show that engineered neutrophil-primed progenitors (NeuPs) derived from human induced pluripotent stem cells (iPSCs) can produce clinically functional neutrophil-like cells (NeuCs), interact with other immune cells to allow them to function rapidly in vivo, and can control a lethal bacterial infection. The number of engineered preservable NeuPs can be efficiently expanded up to 1025-fold, attaining 1014 times more NeuPs than usually used in clinical settings,11 a much higher number than that achieved with current ex vivo manufacturing techniques.12,13 Furthermore, our NeuPs can be differentiated into NeuCs within 4 days. Thus, we can prepare granulocyte products within 4 days whenever patients need granulocyte transfusion therapy (GTX) by expanding or cryopreserving a sufficient amount of NeuPs in advance. This expandable system is much faster than previously reported methods wherein preparation of neutrophils from pluripotent stem cells takes more than 14 days.14-18 NeuPs provide a platform for the robust analyses of human neutrophils and demonstrate that human neutrophils can interact with various types of mouse immune cells to prevent lethal infections.
This system potentially provides a straightforward solution, an off-the-shelf transfusion of neutrophils, for patients suffering from neutrophil dysregulation. This system could provide a platform for the analysis of human neutrophils under various physiological and pathological conditions.
Methods
Human primary CD34+ bone marrow cells and neutrophils
Primary cells were obtained after informed consent. CD34+ bone marrow cells for establishing iPSCs were purchased from Lonza. All studies using human cells were approved by the institutional review boards of the University of Tokyo (approved protocols 2771 and 2314).
Mice
BALB/c mice and NOD.Cg-PrkdcscidIl2rgtm1Wjl/Szj (NSG) mice were purchased from Japan SLC, Inc, and Charles River Laboratories Japan, Inc, respectively. All mice used were aged 8 to 12 weeks. All animal experiments adhered to the University of Tokyo guidelines for animal experiments.
iPSC lines
We used 3 iPSC clones. Bone marrow CD34+ cell-derived iPSCs had been previously established (CD34BM12, CD34BM17).19 Cord blood-derived iPSCs (610B1) were provided by the Center for iPS Cell Research and Application (Kyoto University). The characteristics of iPSC clones are summarized in supplemental Table 1, available on the Blood Web site.
Establishment of expandable NeuPs
iPSC-derived hematopoietic progenitor cells (HPCs)20 were precultured for 12 hours before the initial lentiviral induction of c-Myc and BMI1 under the same culture conditions used at days 0 through 2. The lentiviral induction of BCL-XL was performed from day 10 to day 17. Detailed procedures are provided in supplemental Methods.
Statistical analysis
One-way analysis of variance tests, unpaired 2-tailed t tests, and log-rank test were performed, as indicated in the Figures, using Prism 8 software. The accession number of our RNA sequencing data is GSE156612. Additional methods can be found in the supplemental Methods.
Results
Expandable NeuPs from human iPSCs
Single-cell approaches have provided evidence for lineage commitment, especially neutrophil commitment, of hematopoietic progenitors in mice21-24 and humans.24-26 We established a protocol using short and simple processing to produce NeuPs with sufficient expandability for clinical application and the potential for differentiation into NeuCs. To obtain these expandable NeuPs from integration-free iPSCs (supplemental Figure 1A), we optimized culture conditions using medium supplemented with granulocyte colony stimulating factor (G-CSF; supplemental Figure 1B). To expand progenitors, we used a lentiviral system that employed the Kusabira Orange fluorescent protein as a label for doxycycline (Dox)-induced cassette of c-Myc and BMI1 based on a process initially used to establish expandable megakaryocyte cell lines for ex vivo production of platelets.27,28 Using a culture medium that included stem cell factor, TPO, and FLT-3L followed by gradual replacement with G-CSF, we obtained expandable iPSC-derived progenitors expressing c-Myc and BMI1 (supplemental Figure 1C). The frequency of expandable progenitors in transgene-expressing cells (Kusabira Orange+ cells) expressing c-Myc, BMI1, or both, determined using a limiting dilution assay, was 1/111 to 1/235 (supplemental Figure 1D). Overexpression of c-Myc and BMI1 allowed the progenitors to expand for 10 weeks after induction, and their silencing by culturing in G-CSF in the absence of Dox halted the expansion (supplemental Figure 2A-D). Four days after the silencing, we obtained NeuCs, with banded or segmented nuclei, the expression of the neutrophil-specific surface proteins CD16b and CD66b, and evidence for the presence of the phorbol myristate acetate (PMA)-induced oxidative burst function (supplemental Figure 2E-G), indicating that expandable NeuPs could be generated using this method; however, the expandability was insufficient for clinical application.
To investigate the genetic elements involved in the expandability of engineered NeuPs, we performed single-cell quantitative polymerase chain reaction (sc-qPCR) of transcription factors in the HPCs derived from iPSCs through neutrophil differentiation. We analyzed 77 transcription factors that are enriched among human common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs), promyelocytes/myelocytes, neutrophils, or monocytes based on published data (GSE4988329 and GSE2849230) of HPCs derived from iPSCs through neutrophil differentiation (supplemental Tables 2 and 3). Principal component analysis of sc-qPCR results revealed enrichment of BCL6, CEBPB, CEBPD, and LITAF in a neutrophil-primed subpopulation that became dominant through neutrophil differentiation (supplemental Figure 3A-C). Because CEBPB shares most of its target genes with CEBPA, these findings are compatible with those of previous studies performed at a single-cell resolution, which showed that CEBPA and CEBPD modules are activated in human neutrophil progenitors,26 and that CEBPB and CEBPD are enriched along the human myeloid differentiation trajectory, especially in CMPs and GMPs.31 Of these transcription factors, the overexpression of CEBPB enhanced the expandability of NeuPs for 2 additional weeks, even after preservation for three months at −80°C, but the cells stopped proliferating on the third week after CEBPB induction (supplemental Figure 3D-F). Overexpression of LITAF through neutrophil differentiation apparently accelerated the formation of toxic granules in NeuCs, a response to severe infections by primary neutrophils (supplemental Figure 4A-C). This suggests that preservable NeuPs can be a platform for inducing additional genes to enhance their function and that CEBPB provides some clues to enhance their expandability.
Further analyses were performed to identify factors critical to the expansion of CMPs to GMPs to facilitate the expansion of NeuPs and achieve clinically relevant numbers of cells. CEBPB is a master transcription factor in emergency granulopoiesis, in which hematopoietic progenitors undergo proliferation and differentiation.32CEBPB overexpression, however, did not result in sustained proliferation of NeuPs (supplemental Figure 3F); instead, the cells underwent terminal differentiation presumably because of the action of CEBPB. This did not suit our purpose of eliciting proliferation-promoting activity without differentiation-inducing activity in neutrophil progenitors. CEBPB is involved in accelerating the cell-cycle and inhibiting apoptosis.32,33 Because the CEBPB-induced acceleration of the cell cycle is related to the maintenance of c-Myc, which is already overexpressed in NeuPs (supplemental Figure 2C),32,34 we investigated the effect of enforced CEBPB expression on apoptosis. We analyzed the messenger RNA (mRNA) expression data in a published dataset with a customized gene expression panel (containing 547 immunology-related genes) for CEBPB-induced lymphoid-myeloid transdifferentiation and confirmed that 19 apoptosis-related genes were differentially expressed by enforced CEBPB expression (fold-change > 2.0).35 The analysis of chromatin immunoprecipitation sequencing ENCODE datasets in the K562 myeloid cell line stably expressing CEBPB (GSE91748) indicated that 14 of these 19 apoptosis-related genes were direct targets of CEBPB (supplemental Figure 5A).36 These findings support the notion that enforced CEBPB expression modulates apoptosis, leading to enhanced expandability of NeuPs. These data are also compatible with previous observation that excessive c-Myc expression leads to caspase-dependent apoptosis in a prototype of a megakaryocyte cell line, and that the expression of an antiapoptotic protein, BCL-XL, promotes immortalization of the megakaryocyte cell line.27 In neutrophils, the BCL2 family of proteins inhibits caspase-dependent apoptosis.37,38 We hypothesized that the overexpression of the BCL2 family of proteins does not enhance differentiation but rather the proliferation of NeuPs by blocking apoptosis signaling. Probably, the overexpression of BCL2A1, BCL-XL, and MCL1 minimally perturbs the differentiation from NeuPs to NeuCs because these genes are important for hematopoiesis and myeloid development.39 Consistent with this hypothesis, the overexpression of the BCL2 family of proteins enhanced the expandability of NeuPs, but only BCL-XL supported the expansion of NeuPs in the absence of OP9 feeder cells (supplemental Figure 5B-D). The Dox-inducible overexpression of BCL-XL following the initial induction of c-Myc and BMI1 allowed for the establishment of expandable NeuPs from bone marrow CD34+ cell-derived iPSCs (CD34BM12 and CD34BM17)19 and cord blood-derived iPSCs (610B1, provided by the Center for iPS Cell Research and Application) (Figure 1A).
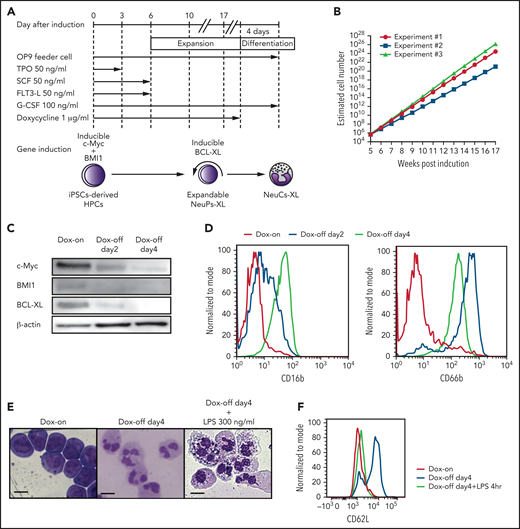
Establishment of expandable neutrophil-primed progenitors enhanced by BCL-XL (NeuPs-XL). (A) A scheme of the establishment of expandable NeuPs-XL from human iPSCs-derived hematopoietic progenitor cells (HPCs) and their differentiation into neutrophil-like cells (NeuCs-XL). (B) Estimated cell number of the expandable progenitors at the presence of doxycycline (Dox). Means of 3 experimental replicates. (C) Protein expression of c-Myc, BMI1, and BCL-XL in the expandable progenitors 2 and 4 days after the withdrawal of Dox and at the presence of Dox. (D) Representative flow cytometry of the neutrophil-specific surface proteins in the expandable progenitors at the presence of Dox (red), 2 (blue), or 4 (green) days after the withdrawal of Dox. (E) Morphology of the expandable progenitors in Wright-Giemsa stain at the presence of Dox or 4 days after the withdrawal of Dox with or without 300 ng/mL lipopolysaccharide (LPS). Scale bar, 10 μm. (F) Representative flow cytometry of CD62L in NeuPs-XL and NeuCs-XL with or without 4-hour LPS stimulation.
Establishment of expandable neutrophil-primed progenitors enhanced by BCL-XL (NeuPs-XL). (A) A scheme of the establishment of expandable NeuPs-XL from human iPSCs-derived hematopoietic progenitor cells (HPCs) and their differentiation into neutrophil-like cells (NeuCs-XL). (B) Estimated cell number of the expandable progenitors at the presence of doxycycline (Dox). Means of 3 experimental replicates. (C) Protein expression of c-Myc, BMI1, and BCL-XL in the expandable progenitors 2 and 4 days after the withdrawal of Dox and at the presence of Dox. (D) Representative flow cytometry of the neutrophil-specific surface proteins in the expandable progenitors at the presence of Dox (red), 2 (blue), or 4 (green) days after the withdrawal of Dox. (E) Morphology of the expandable progenitors in Wright-Giemsa stain at the presence of Dox or 4 days after the withdrawal of Dox with or without 300 ng/mL lipopolysaccharide (LPS). Scale bar, 10 μm. (F) Representative flow cytometry of CD62L in NeuPs-XL and NeuCs-XL with or without 4-hour LPS stimulation.
The inducible overexpression of BCL-XL enhanced the expandability of NeuPs, which reached a density of 1023 cells in a 16-week culture, 1012 times higher than the number of neutrophils required for human transfusion (Figure 1B; supplemental Table 4). Four days after the silencing of c-Myc, BMI1, and BCL-XL, NeuCs expressing neutrophil-specific surface proteins and having segmented nuclei were obtained. Lipopolysaccharide (LPS) stimulation resulted in vacuolation in NeuCs as previously reported40 and in the presence of toxic granules apparently (Figure 1C-E; supplemental Figure 6A). NeuCs also expressed l-selectin (CD62L), which is involved in the initial attachment and rolling of neutrophils, and demonstrated rapid shedding of l-selectin in response to LPS stimulation (Figure 1F). Thus, expandable NeuPs enhanced by the expression of BCL-XL (NeuPs-XL) could be produced on a clinically viable scale to generate NeuCs derived from NeuPs-XL (NeuCs-XL).
Next, we addressed the potential contamination of NeuPs-XL and capacity to revert back to NeuPs-XL after the silencing of inducible genes. A colony-forming capacity assay with Dox readdition in NeuPs-XL after silencing demonstrated that they could revert to the state of NeuPs-XL by Dox readdition within 3 days after Dox removal, but NeuCs-XL are terminally differentiated cells and potential contamination of NeuPs-XL is less than 1/10 000, 4 days after silencing (supplemental Figure 6B-C). When 1.0 × 106 NeuCs-XL were cultured in vitro more than 1 month after removing Dox, the cell number decreased rapidly in a monotone manner after day 4, and no viable cell was detected after day 16 without reexpansion thereafter, which suggests the absence of expandable cells in the Dox-off condition (supplemental Figure 6D). We also addressed the differentiation potential of NeuPs-XL toward other myeloid lineages. Although culturing in G-CSF generated NeuCs-XL exclusively, semisolid culture with multiple cytokines generated large cells without segmented nuclei, suggesting that NeuPs-XL have the potential to differentiate toward other myeloid cells, such as macrophages (supplemental Figure 6E). Next, we examined the effectiveness of simultaneous transduction of 3 genes compared with transduction of c-Myc and BMI1 followed by BCL-XL. Simultaneous overexpression at day 1 after HPC differentiation generated expandable clones. However, 4 days after Dox removal, these cells were differentiated into both neutrophils and macrophages, suggesting that overexpressing BCL-XL earlier cannot generate pure neutrophil progenitors (supplemental Figure 6F).
Long-term culture may impair the function of NeuPs-XL as a result of stem cell senescence. Stem cell senescence is characterized by cell-cycle arrest and the upregulation of several senescence markers such as p16, p18, and p21.41,42 To address the effect of stem cell senescence, we first performed a karyotype analysis at low- and high-passage times (3 and 17 months of culture, respectively) by Giemsa banding. Low-passage NeuPs-XL showed a normal karyotype, whereas high-passage NeuPs-XL had chromosomal abnormalities (supplemental Figure 7A). Although low- and high-passage NeuPs-XL did not exhibit a significant difference in colony-forming capacity, high-passage NeuPs-XL showed a decrease in proliferation, compared with low-passage NeuPs-XL (supplemental Figure 7B-C). Moreover, high-passage NeuPs-XL expressed significantly higher levels of p18 than low-passage NeuPs-XL, whereas p21 levels were similar between low- and high-passage NeuPs-XL (supplemental Figure 7D-E). These findings suggest that long-term culture of NeuPs-XL affects their function as a result of stem cell senescence.
Gene expression profiling of NeuCs-XL
To address whether NeuCs-XL recapitulate the activation of normal neutrophils in terms of gene expression, we performed global gene expression profiling of NeuPs-XL and NeuCs-XL with or without LPS stimulation. Each group in the principal component analysis located as a distinct subpopulation (Figure 2A), and a gene ontology analysis showed that NeuCs-XL had significant changes in the expression levels of genes involved in multiple biological processes, cellular components, and molecular functions as well as in the maturation of neutrophils (supplemental Figure 8A-B). LPS stimulation of NeuCs-XL induced significant changes in biological processes relevant to critical neutrophil functions, including inflammatory and innate immune response, chemokine activity and chemotaxis, cell adhesion, and cytokine activity (Figure 2B). Consistent with RNA-sequencing data, qPCR showed that LPS induced the upregulation of inflammatory cytokines in NeuCs-XL (Figure 2C; supplemental Figure 8C).
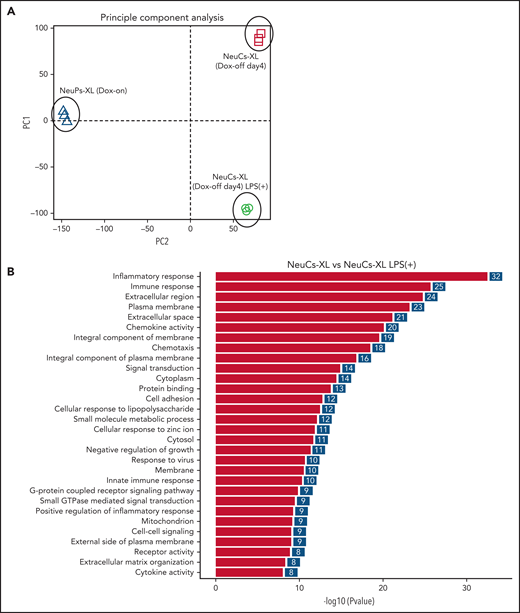
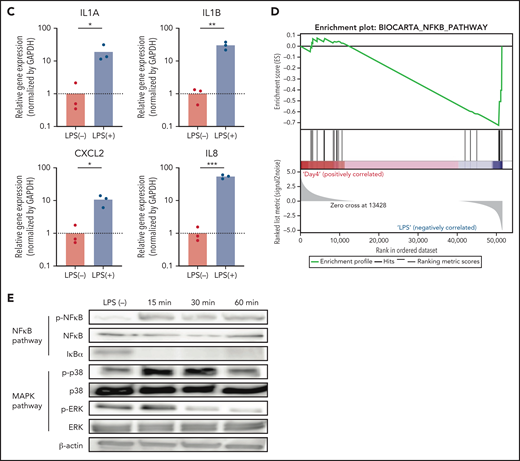
Gene expression profiling in expandable neutrophil-primed progenitors and neutrophil-like cells with or without lipopolysaccharide (LPS) stimulation. (A) Principal component analysis of the gene expression in neutrophil-primed progenitors enhanced by BCL-XL (NeuPs-XL) and neutrophil-like cells from NeuPs-XL (NeuCs-XL) with or without LPS stimulation. (B) Gene ontology analysis of differentially expressed genes in NeuPs-XL with or without 300 ng/mL LPS stimulation. X-axis: -log10(P value) of each term. Y-axis: significant enriched gene ontology term. (C) Gene expression of inflammatory cytokines in NeuCs-XL with or without LPS stimulations, using qPCR. Means of 3 independent experiments, with relative gene expression, normalized to GAPDH expression. Statistical significance was calculated using a Student t test. *P < .05, **P < .01, ***P < .001. (D) Gene set enrichment analysis of the NF-κB pathway in NeuCs-XL with or without LPS stimulation. (E) Phosphorylation and expression of proteins involved in NF-κB and MAPK pathway using NeuCs-XL with 15-, 30-, and 60-minute LPS stimulation or without LPS stimulation.
Gene expression profiling in expandable neutrophil-primed progenitors and neutrophil-like cells with or without lipopolysaccharide (LPS) stimulation. (A) Principal component analysis of the gene expression in neutrophil-primed progenitors enhanced by BCL-XL (NeuPs-XL) and neutrophil-like cells from NeuPs-XL (NeuCs-XL) with or without LPS stimulation. (B) Gene ontology analysis of differentially expressed genes in NeuPs-XL with or without 300 ng/mL LPS stimulation. X-axis: -log10(P value) of each term. Y-axis: significant enriched gene ontology term. (C) Gene expression of inflammatory cytokines in NeuCs-XL with or without LPS stimulations, using qPCR. Means of 3 independent experiments, with relative gene expression, normalized to GAPDH expression. Statistical significance was calculated using a Student t test. *P < .05, **P < .01, ***P < .001. (D) Gene set enrichment analysis of the NF-κB pathway in NeuCs-XL with or without LPS stimulation. (E) Phosphorylation and expression of proteins involved in NF-κB and MAPK pathway using NeuCs-XL with 15-, 30-, and 60-minute LPS stimulation or without LPS stimulation.
We investigated the molecular signaling pathways in NeuCs-XL. A gene set enrichment analysis suggested that LPS induced the activation of the NF-κB pathway, a critical downstream component of LPS stimulation in neutrophils (Figure 2D). Consistent with this result, western blotting showed that LPS stimulation rapidly activated the NF-κB pathway and also resulted in the phosphorylation of p38 in the MAPK pathway, another downstream pathway associated with LPS stimulation, within 15 minutes (Figure 2E). Taken together, these gene expression profiling and signaling pathway analyses indicate that NeuCs-XL have a broad spectrum of functions and rapid molecular responses, which are shared with primary neutrophils.
In vitro functional assay of NeuCs-XL
To evaluate the antibacterial functions of NeuCs-XL, we performed in vitro functional assays to assess adhesion, migration, phagocytosis, bacterial killing, and the formation of neutrophil extracellular traps (NETs). We confirmed not only the high expression of lineage surface proteins but the gene and protein expression of the adhesion molecule, integrin α-M (CD11b), and the chemokine and chemoattractant receptors, CXCR1 and FPR1, in NeuCs-XL (supplemental Figures 6A, 8D, and 9A). We then assessed the dynamics of transcriptional factors critical for neutrophil maturation. Through differentiation, CEBPA, CEBPD, and SPI1 levels increased, whereas those of CEBPE and GFI1 decreased (supplemental Figure 10A). These dynamic changes in transcription factors are compatible with the maturation process of neutrophils.24 Next, we evaluated granule production in NeuCs-XL. NeuCs-XL produced primary (MPO and ELANE), secondary (LTF), and tertiary (MMP9) granules (Figure 3A). Furthermore, the production of primary, secondary, and tertiary granules drastically changed throughout differentiation at the mRNA and protein levels (Figure 3A; supplemental Figure 10B-D). NeuCs-XL were also able to migrate in fetal bovine serum in a dose-dependent manner in a transwell migration assay (Figure 3B). Although NeuCs-XL did not have the ability to migrate in C5a, a potent chemo-attractant for neutrophils, NeuCs-XL did migrate in LTB4 and IL8, significantly (Figure 3C). In a phagocytosis assay using Gram staining, NeuCs-XL phagocytosed Staphylococcus aureus, Escherichia coli, and Candida albicans (Figure 3D; supplemental Figure 10E-F). Flow cytometry confirmed the phagocytosis of FITC-labeled E coli by NeuCs-XL to an extent similar to that of primary human neutrophils in peripheral blood (Figure 3E-F). A PMA-stimulated oxidative burst assay demonstrated that NeuCs-XL also had a PMA-stimulated oxidative burst, which is associated with bacterial killing (Figure 3G-H). We also confirmed NET formation in NeuCs-XL after PMA stimulation (Figure 3I). These results strongly suggest that NeuCs-XL can exert antibacterial activity similar to that of primary human neutrophils.
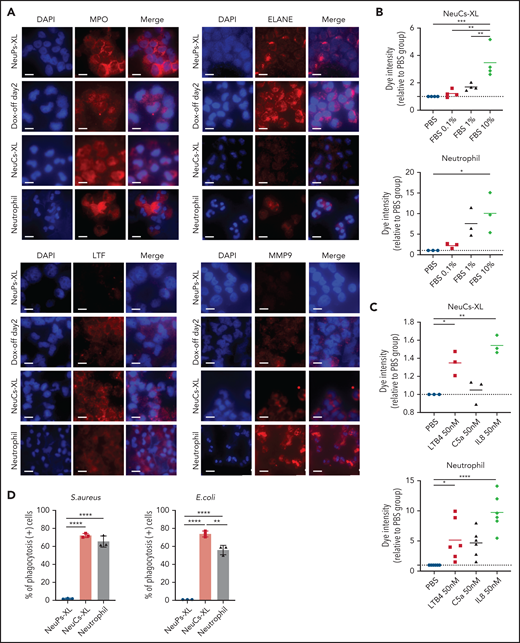
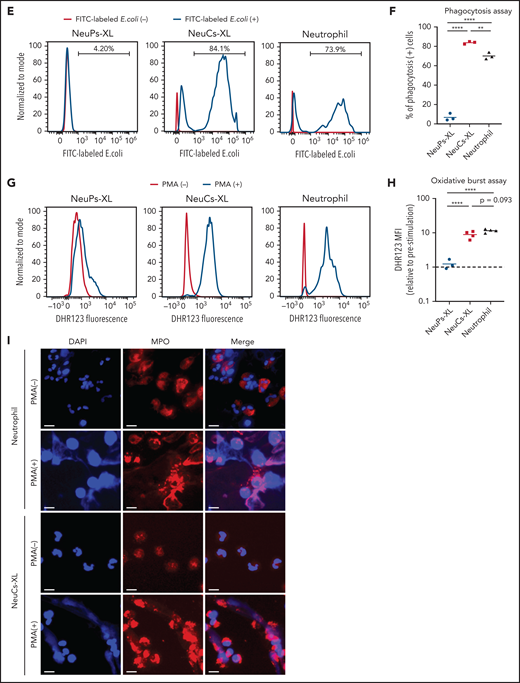
Functional assay of neutrophil-like cells in vitro. (A) Immunocytochemistry of primary (MPO and ELANE), secondary (LTF), and tertiary (MMP9) granules in NeuPs-XL 2 days after removing doxycycline (Dox-off day2), NeuCs-XL, and human primary neutrophils. Scale bar, 10 μm. (B-C) Migration assay of NeuCs-XL and human neutrophils, using a transwell system with 0.1%, 1%, and 10% fetal bovine serum (FBS) or without FBS (B) and with 50 nM LTB4, C5a, and interleukin-8 or without (C). Means of 3 to 6 independent experiments. Statistical significance was calculated using analysis of variance (ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001. (D) The proportion of phagocytosis-positive NeuPs-XL, NeuCs-XL, and human neutrophils incubated with S aureus or E coli. Means of 3 independent experiments. Statistical significance was calculated using ANOVA. **P < .01, ****P < .0001. (E) Representative flow cytometry of phagocytosis assay in NeuCs-XL with or without FITC-labeled E coli. (F) Phagocytosis assay in NeuPs-XL, NeuCs-XL, and human neutrophils. Means of 3 independent experiments. Statistical significance was calculated using ANOVA. **P < .01, ****P < .0001. (G) Representative flow cytometry of oxidative burst assay in NeuPs-XL, NeuCs-XL, and human neutrophils with or without phorbol myristate acetate (PMA) stimulation, using dihydrorhodamine 123. (H) Oxidative burst assay in NeuPs-XL, NeuCs-XL, and human neutrophils using dihydrorhodamine 123. Means of 3 or 4 independent experiments. Statistical significance was calculated using ANOVA. ***P < .001, ****P < .0001. (I) Neutrophil extracellular trap formation of NeuCs-XL and human neutrophils without PMA stimulation or after 4-hour PMA stimulation. Scale bar, 10 μm.
Functional assay of neutrophil-like cells in vitro. (A) Immunocytochemistry of primary (MPO and ELANE), secondary (LTF), and tertiary (MMP9) granules in NeuPs-XL 2 days after removing doxycycline (Dox-off day2), NeuCs-XL, and human primary neutrophils. Scale bar, 10 μm. (B-C) Migration assay of NeuCs-XL and human neutrophils, using a transwell system with 0.1%, 1%, and 10% fetal bovine serum (FBS) or without FBS (B) and with 50 nM LTB4, C5a, and interleukin-8 or without (C). Means of 3 to 6 independent experiments. Statistical significance was calculated using analysis of variance (ANOVA). *P < .05, **P < .01, ***P < .001, ****P < .0001. (D) The proportion of phagocytosis-positive NeuPs-XL, NeuCs-XL, and human neutrophils incubated with S aureus or E coli. Means of 3 independent experiments. Statistical significance was calculated using ANOVA. **P < .01, ****P < .0001. (E) Representative flow cytometry of phagocytosis assay in NeuCs-XL with or without FITC-labeled E coli. (F) Phagocytosis assay in NeuPs-XL, NeuCs-XL, and human neutrophils. Means of 3 independent experiments. Statistical significance was calculated using ANOVA. **P < .01, ****P < .0001. (G) Representative flow cytometry of oxidative burst assay in NeuPs-XL, NeuCs-XL, and human neutrophils with or without phorbol myristate acetate (PMA) stimulation, using dihydrorhodamine 123. (H) Oxidative burst assay in NeuPs-XL, NeuCs-XL, and human neutrophils using dihydrorhodamine 123. Means of 3 or 4 independent experiments. Statistical significance was calculated using ANOVA. ***P < .001, ****P < .0001. (I) Neutrophil extracellular trap formation of NeuCs-XL and human neutrophils without PMA stimulation or after 4-hour PMA stimulation. Scale bar, 10 μm.
NeuCs-XL function in immunodeficient mice
To address the in vivo activity of NeuCs-XL against infection, we first performed in vivo imaging of luciferase2 (luc2)-expressing NeuCs-XL in irradiated immunodeficient NSG mice. The NSG mice were intraperitoneal(ly) (IP) injected with LPS, followed by luc2-expressing NeuCs-XL (Figure 4A). LPS induced inflammation, as determined by an increased white blood cell count in peripheral blood 24 hours after LPS injection (Figure 4B). The LPS-induced inflammation model accumulated NeuCs-XL at the inflammation site within 1 hour, a marked contrast to the diffuse distribution of NeuCs-XL in mice without LPS injection. Persistent survival of injected NeuCs-XL at the inflammation site was also evident (Figure 4C-E).
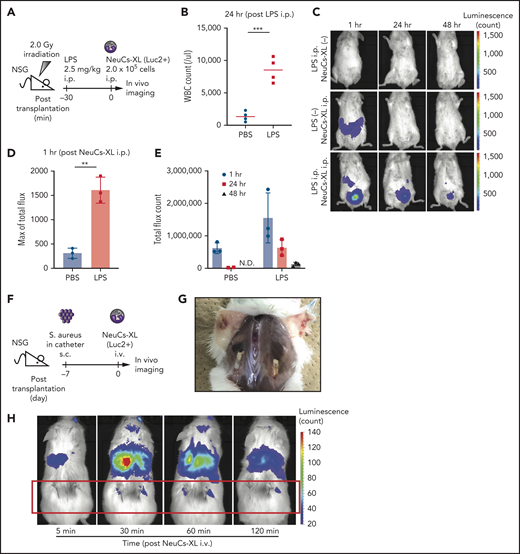
NeuCs-XL accumulate in sites of infection in vivo. (A) A scheme of in vivo imaging of neutrophil-like cells from expandable neutrophil-primed progenitors enhanced by BCL-XL (NeuCs-XL), using Luc2-expressing NeuCs-XL in lipopolysaccharide (LPS)-induced inflammation model. IP injection of LPS was followed by IP injection of Luc2-expressing NeuCs-XL. (B) White blood cell (WBC) count in peripheral blood 24 hours after the IP injection of phosphate-buffered saline (PBS) or LPS. Means of 4 mice in each group. Statistical significance was calculated using a Student t test. ***P = .0004. (C) In vivo imaging of Luc2-expressing NeuCs-XL in LPS-induced inflammation model 1, 24, and 48 hours after their IP injection. (D) Maximum of total flux in the region of interest of mice IP injected with PBS or LPS, followed by Luc2-expressing NeuCs-XL injection. Means ± standard deviation (SD) of 3 mice in each group. Statistical significance was calculated using a Student t test. **P = .0015. (E) Total flux count in the region of interest of mice IP injected with PBS or LPS, followed by Luc2-expressing NeuCs-XL injection. Data were shown 1, 24, and 48 hours after Luc2-expressing NeuCs-XL injection. Means ± SD of 3 mice in each group. ND, not detected. (F) A scheme of in vivo imaging of Luc2-expressing NeuCs-XL and the model of chronic biofilm infection with subcutaneous implantation of the catheters containing S aureus. (G) Catheters containing S aureus were subcutaneously transplanted into both sides of the back in NSG mice. The figure shows chronic infection 7 days after transplantation. (H) In vivo imaging of Lus2-expressing NeuCs-XL in the model of chronic biofilm infection 5, 30, 60, and 120 minutes after IV injection of NeuCs-XL.
NeuCs-XL accumulate in sites of infection in vivo. (A) A scheme of in vivo imaging of neutrophil-like cells from expandable neutrophil-primed progenitors enhanced by BCL-XL (NeuCs-XL), using Luc2-expressing NeuCs-XL in lipopolysaccharide (LPS)-induced inflammation model. IP injection of LPS was followed by IP injection of Luc2-expressing NeuCs-XL. (B) White blood cell (WBC) count in peripheral blood 24 hours after the IP injection of phosphate-buffered saline (PBS) or LPS. Means of 4 mice in each group. Statistical significance was calculated using a Student t test. ***P = .0004. (C) In vivo imaging of Luc2-expressing NeuCs-XL in LPS-induced inflammation model 1, 24, and 48 hours after their IP injection. (D) Maximum of total flux in the region of interest of mice IP injected with PBS or LPS, followed by Luc2-expressing NeuCs-XL injection. Means ± standard deviation (SD) of 3 mice in each group. Statistical significance was calculated using a Student t test. **P = .0015. (E) Total flux count in the region of interest of mice IP injected with PBS or LPS, followed by Luc2-expressing NeuCs-XL injection. Data were shown 1, 24, and 48 hours after Luc2-expressing NeuCs-XL injection. Means ± SD of 3 mice in each group. ND, not detected. (F) A scheme of in vivo imaging of Luc2-expressing NeuCs-XL and the model of chronic biofilm infection with subcutaneous implantation of the catheters containing S aureus. (G) Catheters containing S aureus were subcutaneously transplanted into both sides of the back in NSG mice. The figure shows chronic infection 7 days after transplantation. (H) In vivo imaging of Lus2-expressing NeuCs-XL in the model of chronic biofilm infection 5, 30, 60, and 120 minutes after IV injection of NeuCs-XL.
Next, we IV-injected luc2-expressing NeuCs-XL into irradiated NSG mice (supplemental Figure 11A). The number of injected NeuCs-XL was 1.0 × 107 cells (5.0 × 108 cells/kg), almost equivalent to the titer used for granulocyte transfusion in humans. Although most IV-injected NeuCs-XL were trapped in the lung, a portion circulated in the peripheral blood (supplemental Figure 11B-D). Intravenously injected NeuCs-XL were no longer detectable by flow cytometry of peripheral blood or by in vivo imaging 2 days after their injection (supplemental Figure 11B-D). These findings suggest that NeuCs-XL have a short lifespan in vivo, which could be safe for clinical application. To address the distribution of IV-injected NeuCs-XL, we performed in vivo imaging 60 and 120 minutes after injection of NeuCs-XL with or without IP LPS injection (supplemental Figure 11E). Initially, NeuCs-XL were mainly trapped in the lungs, and then distributed to the liver and spleen in the LPS− group. Contrarily, in the LPS+ group, some NeuCs-XL distributed to the LPS-induced inflammation site rather than to the liver or spleen (supplemental Figure 11F-G).
To address concerns about potential tumorigenicity of NeuCs-XL in vivo, we followed 6 irradiated NSG mice IV injected with NeuCs-XL for 8 weeks (supplemental Figure 12A). In fluorescence-activated cell sorting (FACS) analysis of peripheral blood and whole-body in vivo imaging, NeuCs-XL were not detectable in any of the 6 mice 2 weeks after the initial injection (supplemental Figure 12B-D). In addition, we followed 6 irradiated NSG mice for 4 weeks in the condition of inflammation and the presence of Dox (supplemental Figure 12E) and confirmed that NeuCs-XL were not detectable in any of the 6 mice 2 weeks after the initial injection (supplemental Figure 12F-H).
Moreover, we assessed the cell fate of IV-injected NeuPs-XL following G-CSF administration (supplemental Figure 13A). IV-injected NeuPs-XL remained detectable in the liver to some extent by in vivo imaging 4 days after their injection (supplemental Figure 13B), but human CD45+ cells were almost undetectable by FACS in peripheral blood or bone marrow (supplemental Figure 13C-D), which suggests that injecting NeuCs-XL is the optimal method.
Next, we performed in vivo imaging of luc2-expressing NeuCs-XL in a chronic biofilm infection model using S aureus,43 Luc2-expressing NeuCs-XL were IV injected into immunodeficient NSG mice that had been subcutaneously transplanted with catheters precolonized by S aureus into both sides of their back (Figure 4F-G). This chronic biofilm infection model demonstrated that NeuCs-XL promptly migrated to the infection site within 30 minutes after IV injection (Figure 4H). These findings suggest that NeuCs-XL can readily accumulate at the site of inflammation and infection, and therefore provide a platform for the robust analysis of human neutrophils in vivo.
NeuCs-XL recruit mouse immune cells and protect from lethal infection
To address the interaction between NeuCs-XL and other types of immune cells, we established an experimental system using a lethal acute infection model. The IP injection of S aureus induced acute lethal peritonitis in immunocompetent BALB/c mice (supplemental Figure 14A). We confirmed the colony-forming unit (CFU)-dependent lethality during the 24 hours after its injection and determined the lethal CFU dose under normal conditions and after radiation-induced leukopenia (supplemental Tables 5 and 6). The IP injection of NeuCs-XL in the acute peritonitis model under normal conditions (immunocompetent BALB/c mice administered a lethal dose of S aureus) improved survival in a dose-dependent manner and a large enough dose of NeuCs-XL achieved complete survival as well as human neutrophils (1.0 × 107 NeuCs-XL: 10/10, 1.0 × 107 human neutrophils: 3/3, respectively) (supplemental Figure 14B). Furthermore, we confirmed that the IP injection of NeuCs-XL decreased the CFU of S aureus in the peritoneal cavity 16 hours after injection (supplemental Figure 14C), suggesting that NeuCs-XL injection protected from lethal infection.
To evaluate the function of the injected neutrophils more clearly, we performed neutrophil depletion with an anti-Ly6G antibody, avoiding the possible effects of host murine neutrophils (Figure 5A; supplemental Figure 14D). S aureus peritonitis after neutrophil depletion resulted in about 85% (6 of 7) lethality and the injection of mouse neutrophils improved the survival of acute peritonitis (Figure 5B), which provides a proof of concept in this experimental model that IP-injected neutrophils function to prevent lethal infection. Notably, the injection of NeuCs-XL achieved complete survival, whereas the injection of NeuPs-XL did not show any effect on survival compared with the injection of PBS (Figure 5B). Furthermore, primary human neutrophils had a similar significance on survival compared with mouse neutrophils (Figure 5B). These findings indicate that the improvement of survival is due to the neutrophil function of NeuCs-XL, and is not just an immunological response against the xenograft.


NeuCs-XL recruit murine immune cells and protect from lethal infection. (A) A scheme of survival analysis in the model of lethal bacterial peritonitis with S aureus. (B) Kaplan-Meier survival curve of neutrophil-depleted BALB/c mice with bacterial peritonitis, followed by the IP injection of NeuPs-XL, NeuCs-XL, murine neutrophils, human neutrophils, or PBS. Dotted lines indicate 95% confidence interval. Statistical significance was calculated using log-rank test. *P < .05. (C) Gene expression of chemokines and cytokines in NeuPs-XL and NeuCs-XL with or without S aureus stimulation, using qPCR. Means of 3 independent experiments, with relative gene expression, normalized to GAPDH expression. Statistical significance was calculated using ANOVA. *P < .05, **P < .01, ***P < .001, ****P < .0001. (D) A scheme of FACS analysis of peritoneal exudated cells in the model of bacterial peritonitis with S aureus. (E) Cell numbers of peritoneal exudate cells (total cells, dendritic cells, and macrophages) in each condition. Means of 3 to 6 independent experiments. Statistical significance was calculated using ANOVA. *P < .05, **P < .01, ***P < .001. (F) A scheme of FACS analysis of peritoneal exudated cells in the model of bacterial peritonitis with S aureus using TLR-2 blocked NeuCs-XL. (G) Cell numbers of peritoneal exudate cells (human CD45+ cells, dendritic cells, and macrophages) in each condition. Statistical significance was calculated using a Student t test. *P < .05. (H) A scheme of survival analysis in the model of lethal bacterial peritonitis with S aureus after sublethal irradiation. (I) Kaplan-Meier survival curve of irradiated BALB/c mice with bacterial peritonitis, followed by the IP injection of NeuCs-XL or PBS. Statistical significance was calculated using log-rank test. **P = .0062.
NeuCs-XL recruit murine immune cells and protect from lethal infection. (A) A scheme of survival analysis in the model of lethal bacterial peritonitis with S aureus. (B) Kaplan-Meier survival curve of neutrophil-depleted BALB/c mice with bacterial peritonitis, followed by the IP injection of NeuPs-XL, NeuCs-XL, murine neutrophils, human neutrophils, or PBS. Dotted lines indicate 95% confidence interval. Statistical significance was calculated using log-rank test. *P < .05. (C) Gene expression of chemokines and cytokines in NeuPs-XL and NeuCs-XL with or without S aureus stimulation, using qPCR. Means of 3 independent experiments, with relative gene expression, normalized to GAPDH expression. Statistical significance was calculated using ANOVA. *P < .05, **P < .01, ***P < .001, ****P < .0001. (D) A scheme of FACS analysis of peritoneal exudated cells in the model of bacterial peritonitis with S aureus. (E) Cell numbers of peritoneal exudate cells (total cells, dendritic cells, and macrophages) in each condition. Means of 3 to 6 independent experiments. Statistical significance was calculated using ANOVA. *P < .05, **P < .01, ***P < .001. (F) A scheme of FACS analysis of peritoneal exudated cells in the model of bacterial peritonitis with S aureus using TLR-2 blocked NeuCs-XL. (G) Cell numbers of peritoneal exudate cells (human CD45+ cells, dendritic cells, and macrophages) in each condition. Statistical significance was calculated using a Student t test. *P < .05. (H) A scheme of survival analysis in the model of lethal bacterial peritonitis with S aureus after sublethal irradiation. (I) Kaplan-Meier survival curve of irradiated BALB/c mice with bacterial peritonitis, followed by the IP injection of NeuCs-XL or PBS. Statistical significance was calculated using log-rank test. **P = .0062.
Neutrophils express several cytokines and chemokines after activation and induce an inflammatory response via other immune cells in addition to their direct bactericidal capacity. To assess the functional interaction between NeuCs-XL and other types of endogenous immune cells, we evaluated the expression of cytokines and chemokines after activation of NeuCs-XL by S aureus. NeuCs-XL expressed multiple cytokines and chemokines at levels tens to hundreds of times higher than NeuPs-XL in a steady state (Figure 5C). Moreover, NeuCs-XL exposure to S aureus in vitro led to the upregulation of cytokines and chemokines at the mRNA and protein levels, whereas NeuPs-XL did not show the same responses (Figure 5C; supplemental Figure 14E; supplemental Table 7). To confirm this function in vivo, we analyzed the exudate cells in the peritoneal cavity (Figure 5D) and found that NeuCs-XL injection under infection conditions significantly recruited multiple types of murine immune cells, including dendritic cells and macrophages, and that only NeuCs-XL remained at the infection site (Figure 5E; supplemental Figure 15A-D). To clarify whether the immune response of NeuCs-XL against S aureus recruits endogenous immune cells, we blocked TLR2 signaling in NeuCs-XL with an anti-human TLR2 antibody (Figure 5F). Blocking TLR2 did not affect the number of viable NeuCs-XL, but significantly reduced the recruitment of dendritic cells and macrophages (Figure 5G; supplemental Figure 15E-F). Finally, we used sublethally irradiated mice to replicate the clinical situation of severe leukopenia in which endogenous immune cells are quite limited (Figure 5H; supplemental Figure 15G). Under irradiation-induced severe leukopenia, NeuCs-XL injection significantly elongated the survival time (Figure 5I), further demonstrating their efficacy. Collectively, our findings provide compelling evidence that high doses of NeuCs-XL protect from lethal infection and interact with endogenous immune cells in mice.
Discussion
About a half-century has passed since GTX was first established,44 but obtaining an adequate number of neutrophils for transfusion has remained a critical problem.11,45-47 Although a higher dose of granulocytes for transfusion might be promising, previous randomized controlled trials have not conclusively demonstrated the efficacy of high-dose GTX, at least partially, because of the failure to collect a sufficient number of granulocytes from healthy donors.11,48 We demonstrate that expandable and preservable NeuPs-XL can be generated at cell densities beyond the scale required for clinical application and that these cells effectively ameliorate bacterial infection, an essential quality for off-the-shelf transfusion therapies. Although serum-free, feeder-free, and transgene-free generation of NeuP should be required for clinical use and be addressed in future studies, we believe that our findings provide a significant impact on mass production of neutrophils at a clinically relevant scale. A complementary approach to the use of neutrophils in the clinic is the transfusion of myeloid progenitor cells, which has significant impact on preventing infections.49,50 Therefore, improving the efficacy of engraftment and expansion of NeuPs-XL in vivo for preventing febrile neutropenia is another important future challenge.
We show that NeuCs-XL have several fundamental neutrophil functions namely adhesion, migration, phagocytosis, bactericidal capacity, and NET formation. Our in vivo experiment is artificial; thus, this result does not directly prove the efficacy of GTX in clinical settings, but strongly suggests the positive effect of NeuCs-XL against infection in vivo. Besides acquiring these functions to attack infections by themselves, NeuCs-XL could interact with other immune cells to amplify both innate and acquired immune response. Experimental models in vivo to assess immunological interactions have been, to our knowledge, quite limited for human neutrophils, mainly because of the low quality and quantity of human neutrophils available, and the lack of animal models. Herein, the establishment of NeuPs-XL and an acute lethal peritonitis model followed by IP injection of neutrophils allowed us to perform a robust analysis of the interaction between injected neutrophils and other host immune cells. Notably, we demonstrate that human neutrophils recruit various types of mouse immune cells to improve the immune response.
There are multiple limitations in this study, especially in the analyses of immunological interactions. First, the IP injection of neutrophils is artificial. However, we do have a proof of concept of this infection model because we demonstrated that intraperitoneally injected primary murine neutrophils improved survival. Moreover, we demonstrated that intravenously injected NeuCs-XL promptly migrated to the infection site within 30 minutes in NSG mice. Given that one of the features of NeuPs-XL is expandability, we can feasibly apply a sufficient number of IV-injected NeuCs-XL for infections if NeuCs-XL are trapped less in the lung. Second, to analyze multiple types of immune cells including lymphoid cells, we used immunocompetent mice. In immunocompetent mouse models, immune responses for xenograft should occur and could be an unavoidable bias in the analyses of immunological interactions. However, because injection of NeuPs-XL did not have any significant effect on survival or the recruitment of host mouse immune cells, it would be reasonable to conclude that NeuCs-XL interact specifically with mouse immune cells and do not provoke an immune response against the xenograft, thereby contributing to the control of lethal infection. It also supports the interaction of NeuCs-XL with murine immune cells that NeuCs-XL injection elongates survival time under severe leukopenia in which endogenous murine immune cells are quite limited, rather than providing full survival as would be seen under normal conditions. Given that some cytokines and chemokines involved in the recruitment of immune cells have been reported to have cross-reactivity between humans and mice,51,52 cytokines and chemokines secreted from human NeuCs-XL could recruit and activate murine immune cells to prevent acute lethal peritonitis. Third, issues of HLA disparity should be considered before clinical application. In clinical settings, matching HLA for GTX is not required, and a report from the National Institutes of Health describing 32 GTX cases of aplastic anemia showed that HLA alloimmunization status was not associated with mean posttransfusion absolute neutrophil count, and transfusion-related acute lung injury did not occur.53 However, there is a possibility that HLA disparity may cause unfavorable immune reaction and impair effectiveness of GTX to some extent. To deal with this problem, depleting HLA class I by β2-microglobulin knock-out54 or matching HLA types using HLA homozygous iPSC lines55 should be considered in the future.
Overall, our study demonstrates that NeuCs-XL share many features of neutrophils in terms of functions including interaction with various types of immune cells, global gene expression profile, and response to chemicals and microorganisms. These findings strongly suggest that NeuPs-XL provide insights into a broad spectrum of neutrophil functions involved in innate and adaptive immunity, including injury, autoimmunity, and cancer.
Acknowledgments
The authors thank Chad Cowan of Harvard University for complementary DNA of CCAAT enhancer binding protein β (CEBPB); the ENCODE consortium, and the Michael Snyder laboratory at Stanford University for generating the chromatin immunoprecipitation sequencing (GSE91748); Arika Nukina-Shimura for experimental advice; Yoko Hokama, Keiko Tanaka, and Yoshiko Ito for expert technical assistance; and Kyowa Kirin Co Ltd for recombinant human G-CSF.
This work was funded by the Center of Innovation Program (Japan Science and Technology Agency, grant 10101519), the Japan Agency for Medical Research and Development (grant 18072499), and joint research with Kyowa Kirin Co Ltd.
Authorship
Contribution: M.M. designed the research, performed experiments, supervised experiments, analyzed data, and wrote the manuscript; Y. Ito designed the research, performed experiments, analyzed data, and wrote the manuscript; F. Nakahara, J.K., S.A., and Y.K. designed the research and supervised experiments; F. Nakamura, T.H., Y. Iwasaki, and T.K. performed experiments and analyzed data; A.Y. conceptualized and designed the research and supervised experiments; and M.K. conceptualized and designed the research, supervised experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: M.M., S.A., and M.K. have received research funding from Kyowa Kirin Co. Ltd. Y. Iwasaki and T.K. were employed by Kyowa Kirin Co. Ltd. while engaged in this research project. M.M., Y. Ito, and M.K. are listed as inventors on patents describing methods for generating iPSC-derived engineered neutrophils and their functions.
Correspondence: Mineo Kurokawa, 7-3-1 Hongo, Bunkyo-City, Tokyo 113-8655, Japan; e-mail: kurokawa-tky@umin.ac.jp.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal