


Abstract
The BCL6 corepressor (BCOR) is a transcription factor involved in the control of embryogenesis, mesenchymal stem cells function, hematopoiesis, and lymphoid development. Recurrent somatic clonal mutations of the BCOR gene and its homolog BCORL1 have been detected in several hematologic malignancies and aplastic anemia. They are scattered across the whole gene length and mostly represent frameshifts (deletions, insertions), nonsense, and missence mutations. These disruptive events lead to the loss of full-length BCOR protein and to the lack or low expression of a truncated form of the protein, both consistent with the tumor suppressor role of BCOR.BCOR and BCORL1 mutations are similar to those causing 2 rare X-linked diseases: oculofaciocardiodental (OFCD) and Shukla-Vernon syndromes, respectively. Here, we focus on the structure and function of normal BCOR and BCORL1 in normal hematopoietic and lymphoid tissues and review the frequency and clinical significance of the mutations of these genes in malignant and nonmalignant hematologic diseases. Moreover, we discuss the importance of mouse models to better understand the role of Bcor loss, alone and combined with alterations of other genes (eg, Dnmt3a and Tet2), in promoting hematologic malignancies and in providing a useful platform for the development of new targeted therapies.
Introduction
The BCL6 corepressor (BCOR) is a tumor suppressor gene that was first identified in a 2-hybrid screen for interactors with the POZ domain of the transcriptional repressor BCL6.1 Its product is a nuclear protein involved in lymphoid development2 (potentiating BCL6 repression1), maintaining pluripotency of human embryonic stem cells,3,4 and regulating mesenchymal stem cells function5 and hematopoiesis.6 The BCL6 corepressor-like protein 1 (BCORL1)7 shares several features with BCOR but also shows distinctive characteristics, suggesting it may play different functions.8
In 2011, searching for new mutations in de novo adult acute myeloid leukemia (AML), we applied whole exome sequencing to AML patients with normal karyotype lacking NPM1, CEBPA, FLT3-ITD, IDH1, and MLL-PTD (the only known mutations at that time), which led us to discovery somatic clonal BCOR mutations in AML.9 Another group simultaneously identified mutations of the BCOR homolog BCORL1 in de novo and secondary AML.10 Since then, mutations of these genes were increasingly reported in both malignant and nonmalignant hematologic diseases.
Germinal BCOR mutations also cause oculofaciocardiodental (OFCD) syndrome, a rare X-linked dominant disease that is embryonic lethal in males.11 OFCD syndrome is characterized by congenital cataracts, abnormal facial traits, cardiac defects and dental anomalies, including canine teeth with extremely long roots.11BCOR mutations were also identified in Lenz microphthalmia syndrome.11,12 The great variability in the severity of OFCD syndrome among females is likely due to differences in the proportion of cells (mosaicism) carrying a transcriptionally active X chromosome with BCOR mutation in various tissues.13 In fact, BCOR is located on chromosome X, and X inactivation occurs randomly in the early embryo. Alternatively, a phenotype may not become manifest if cells harboring the BCOR mutation on the active X chromosome fail to survive (eg, leukocytes from OFCD syndrome patients show 96% to 100% allelic skewing in favor of cells expressing wild-type BCOR).13 Germline BCORL1 mutations cause the Shukla-Vernon syndrome (named SHUVER), an X-linked recessive disorder characterized by global developmental delay, variably impaired intellectual development and behavioral abnormalities.14,15
Although BCOR mutations are relatively uncommon, their occurrence has been increasingly reported in various hematologic diseases. The scope of this review is to bring all information in a single place, so that it can serve as reference for researchers and clinicians dealing with this issue. In particular, here we focus on the structure and function of normal BCOR in normal hematopoietic and lymphoid tissues and review the frequency and clinical significance of BCOR mutations in hematologic diseases. When available, data on BCORL1 homolog are also provided. Moreover, we discuss the importance of mouse models to better understand the role of Bcor loss in promoting hematologic malignancies and providing a platform for developing new targeted therapies.
BCOR and BCORL1 genes and proteins
The characteristics of BCOR and BCORL1 are summarized in Table 1. The BCOR gene locates at position 11.4 of the short arm of chromosome X and is made up of 15 exons1 (supplemental Figure 1, available on the Blood Web site). BCOR shows a nuclear localization16 that is driven by the interaction of its 2 nuclear localization signals17 with the nuclear import proteins KPNA2, 4, and 6.18 The BCOR protein is regarded to be widely expressed in the lympho-hematopoietic system,19 but immunohistochemical studies with specific monoclonal antibodies are missing. Several alternative spliced transcript variants encoding 4 different isoforms (a, b, c, d) have been described.1,20 The main isoform c uses 15 exons to generate a protein of 1755 amino acids (192 kDa).1 Only a few isoforms retain known protein interactions, depending on the domains preserved by alternative splicing.
Characteristics of BCOR and BCORL1 genes and proteins
| Characteristics | BCOR gene | BCORL1 gene |
|---|---|---|
| Location | Chromosome X (band Xp11.4) | Chromosome X (band Xp26.1) |
| No. of exons | 15 | 13 |
| Associated genetic syndrome | OFCD | Shukla-Vernon (SHUVER) |
| Mutations | Frameshifts, nonsense, and missense* | Frameshifts, nonsense, and missense*,† |
| Translocations | Rare (APL)* | No |
| ITD of PUFD (solid tumors) | Yes | No |
| Characteristics | BCOR protein | BCORL1 protein |
| Length | 1755 amino acids | 1711 amino acids |
| Subcellular location | Nucleosol and nuclear dots of various size | Speckle-like nuclear dots of consistent size |
| Major protein domains | BCL6-binding domain, PUFD motif, MLLT3-binding domain Tandem ankyrin repeats | CtBP1-binding site, PUFD motif, 2 LXXLL motifs, tandem ankyrin repeats |
| PUFD motif | Disordered | Ordered |
| Function | Transcriptional corepressor | Transcriptional corepressor |
| Interactors | BCL6, HDACs class I and II, MLLT3, FXBL10/JHDM1B, MLLT1/ENI, ZBTB5, SP1, ZBTB2, ZBTB7A/Pokemon, PCGF1, RING1A/B KDM2D | HDACs class I and II, CTBP1, PCGF1 |
| Characteristics | BCOR gene | BCORL1 gene |
|---|---|---|
| Location | Chromosome X (band Xp11.4) | Chromosome X (band Xp26.1) |
| No. of exons | 15 | 13 |
| Associated genetic syndrome | OFCD | Shukla-Vernon (SHUVER) |
| Mutations | Frameshifts, nonsense, and missense* | Frameshifts, nonsense, and missense*,† |
| Translocations | Rare (APL)* | No |
| ITD of PUFD (solid tumors) | Yes | No |
| Characteristics | BCOR protein | BCORL1 protein |
| Length | 1755 amino acids | 1711 amino acids |
| Subcellular location | Nucleosol and nuclear dots of various size | Speckle-like nuclear dots of consistent size |
| Major protein domains | BCL6-binding domain, PUFD motif, MLLT3-binding domain Tandem ankyrin repeats | CtBP1-binding site, PUFD motif, 2 LXXLL motifs, tandem ankyrin repeats |
| PUFD motif | Disordered | Ordered |
| Function | Transcriptional corepressor | Transcriptional corepressor |
| Interactors | BCL6, HDACs class I and II, MLLT3, FXBL10/JHDM1B, MLLT1/ENI, ZBTB5, SP1, ZBTB2, ZBTB7A/Pokemon, PCGF1, RING1A/B KDM2D | HDACs class I and II, CTBP1, PCGF1 |
Sometimes may co-occur in AML and MDS.
Rare variant of acute promyelocytic leukemia (APL).
The BCOR protein contains 3 well-established functional domains, whereas the significance of the C-terminal tandem ankyrin (ANK) repeats remains unknown (Figure 1). The BCL6 binding site located at the N terminus of BCOR interacts with the POZ domain of BCL61,21 and with the α-helical region of interferon regulatory factor 8 (also targeting BCL6).22
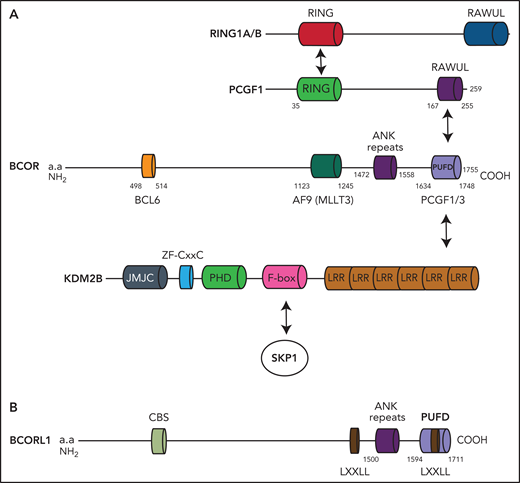
BCOR and BCORL1 proteins. (A) The BCOR protein is characterized by the BCL6 binding site, the AF9 (MLLT3) binding site, the ANK repeats, and the PUFD binding site capable to dimerize with PCGF1. When the BCOR PUFD domain binds to the RAWUL domain of PCGF1, the complex acquires stability and therefore BCOR is able to interact with the leucine-rich repeat domains of KDM2B. Other components of the multiprotein complex include the catalytic enzyme RING1A/B, RYPB, and SKP1. (B) The BCORL1 protein is characterized by the CtBP1 binding site (CBS), 2 LXXLL (nuclear receptor recruitment motifs), the ANK repeats, and the PUFD binding site.
BCOR and BCORL1 proteins. (A) The BCOR protein is characterized by the BCL6 binding site, the AF9 (MLLT3) binding site, the ANK repeats, and the PUFD binding site capable to dimerize with PCGF1. When the BCOR PUFD domain binds to the RAWUL domain of PCGF1, the complex acquires stability and therefore BCOR is able to interact with the leucine-rich repeat domains of KDM2B. Other components of the multiprotein complex include the catalytic enzyme RING1A/B, RYPB, and SKP1. (B) The BCORL1 protein is characterized by the CtBP1 binding site (CBS), 2 LXXLL (nuclear receptor recruitment motifs), the ANK repeats, and the PUFD binding site.
The polycomb group RING finger (PCGF) Ub-like fold discriminator (PUFD) binding site is located at the C terminus of BCOR and interacts with the PCGF homolog 1 (PCGF1)23 (Figure 1). BCOR, the PCGF1/RING enzymatic core, and KDM2B are critical components of the noncanonical polycomb repressive complex PRC1.1.24-27 (Figure 2). The noncanonical PRC1.1 complex contains other proteins that may be tissue specific, for example, in germinal center B cells, BCOR forms a noncanonical PRC1.1 complex containing BCL6 and the CBX8 subunit.2,24,26 This interaction allows recruitment of the complex to specific chromatin regions via mechanisms involving interaction with both chromatin marks and sequence-specific transcription factors. Finally, a BCOR-independent mechanism of recruitment of PRC2/canonical PRC1 complexes to nonresponsive targets that may counteract the gene activation because of BCOR loss has been previously described.3
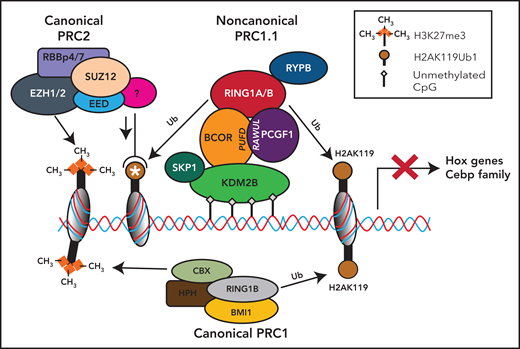
Noncanonical PRC1.1 complex and canonical PRC2 and PCR1 complexes in HSCs. The BCOR complex is recruited to the chromatin via binding of KDM2B to nonmethylated CpG islands, and it catalyzes the ubiquitination of the histone H2A at Lys119 (H2AK119ub) via the RING-PCGF1 enzymatic core. Ubiquinated loci (white asterisk) recruit the histone methyltransferase EZH2, one of the components of the polycomb repressor complex 2 (PRC2). PRC2 is then responsible for the histone H3 methylation at Lys27 (H3K27me3). All these histone modifications lead to the suppression of gene transcription. Canonical PRC1 complex through its components RING1B and CBX catalyzes both the ubiquitination of the histone H2A at Lys119 (H2AK119ub) and the histone H3 methylation at Lys27 (H3K27me3), also leading to the suppression of gene transcription.
Noncanonical PRC1.1 complex and canonical PRC2 and PCR1 complexes in HSCs. The BCOR complex is recruited to the chromatin via binding of KDM2B to nonmethylated CpG islands, and it catalyzes the ubiquitination of the histone H2A at Lys119 (H2AK119ub) via the RING-PCGF1 enzymatic core. Ubiquinated loci (white asterisk) recruit the histone methyltransferase EZH2, one of the components of the polycomb repressor complex 2 (PRC2). PRC2 is then responsible for the histone H3 methylation at Lys27 (H3K27me3). All these histone modifications lead to the suppression of gene transcription. Canonical PRC1 complex through its components RING1B and CBX catalyzes both the ubiquitination of the histone H2A at Lys119 (H2AK119ub) and the histone H3 methylation at Lys27 (H3K27me3), also leading to the suppression of gene transcription.
The PUFD termini of BCOR, which are critical for binding to the ubiquitin-like RAWUL domain of PCGF1, are structurally disordered and become ordered only upon binding PCGF128 (Figure 1). In this way, the PCGF1/BCOR PUFD terminal residues are placed in conformations that are required to interact with the leucine-rich repeats of KDM2B (Figure 1).28
The PRC1.1 complex then is recruited to unmethylated cytosine guanine dinucleotide (CpG) islands that are frequently located around transcription start sites. Binding to unmethylated CpG islands occurs through the zinc finger-CxxC (ZF-CxxC) DNA-binding domain of histone demethylase KDM2B3,29 (Figure 2) that specifically demethylates H3K36me2 via its jmjC domain. Binding of PCGF1-BCOR complex with KDM2B stimulates the E3 ligase activity of RING1B that in turn monoubiquitylates H2A on K119, promoting the accrual of canonical PRC2 complex30 to monoubiquinated loci.26 Conversely, BCOR loss results into a decrease of H2AK119ub1 at promoter regions of Hoxa and Cebpa family genes.31 Thus, BCOR appears critical to couple the RING-PCGF1 enzymatic core to the chromatin bound KDM2B subunit. This function may be disrupted in case of BCOR loss or truncation. The tissue specificity of the PRC1.1 complex may account for the localized rather than global effects of BCOR loss on H2A ubiquitination on K119.
EZH2, 1 of the members of the PRC2 complex, mediates the mono-, di-, and trimethylation of lysine 27 of histone H3 to generate H3K27me1/me2/me3 (Figure 2).6 These events ultimately lead to repression of transcription by histone modifications in specific promoter regions (Figure 2). Conversely, the Kdm2b depletion (eg, in mouse embryonic stem cells) induces the derepression of lineage-specific genes and early differentiation.32
The third functional binding site of BCOR directly interacts with the transcriptional regulator AF9 (MLLT3),33 the common fusion partner of mixed lineage leukemia (MLL) in leukemias.34 In particular, AF9 binds the 2 BCOR isoforms with a unique 34 amino acid sequence in the midportion of the protein.33
The BCORL1 gene maps to chromosome Xq25-q26.1.7 The encoded nuclear protein seems to be expressed at higher levels in testis and prostate than in other tissues7 (Table 1). BCORL1 is 1711 amino acids long and contains a PUFD domain necessary and sufficient to bind PCGF1 RAWUL and together bind KDM2B35 (Figure 1). However, unlike BCOR, it lacks the BCL6 and MLLT3 binding sites and contains an LXXLL nuclear receptor recruitment motif and a PXDLS motif that interacts with the C-terminal binding protein (CtBP) corepressor, resulting in negative regulation of its target genes, including E-cadherin (Figure 1; Table 1). The repressive BCORL1 activity is mediated at least partially by class II histone deacetylases7 (Table 1).
BCOR function in hematopoiesis and lymphoid development
The BCOR-containing PRC1.1 complex regulates hematopoiesis by opposing differentiation toward the myeloid lineage6,31,36,37 through repression of HoxA and Cebp family genes.27,38 Conversely, after depletion of Pcgf139 or Kdm2b,40 hematopoietic stem cells (HSCs) are biased toward the myeloid lineage. Moreover, myeloid cells from mice lacking BCOR exons 9 to 10 and expressing a C terminus truncated BCOR unable to bind Pcgf1 show higher proliferation and differentiation rates in vitro.37 Myeloid-biased hematopoiesis is also found in BcorDE9-10/y progenitors.31 Moreover, depletion of both Runx1 and Pcgf1 sustain the proliferative status and perturb the differentiation of HSCs because of increased expression of Hoxa9.38 Thus, the BCOR-containing PRC1.1 complex is required to repress myeloid regulatory genes and to commit progenitors toward lymphopoiesis. Accordingly, loss-of-function Bcor leads to a selective disadvantage in B- and T-cell lineages.40
BCL6 is strongly expressed by the germinal center B cells of lymphoid follicles,41,42 and by interacting with BCOR recruits the PRC1.1 complex that leads to the epigenetic transcriptional repression of BCL6 target genes (Figure 3). Specifically, genes controlling differentiation of B cells to plasma cells (PRDM1, IRF4) and cell cycle checkpoint (CDKN1A, CDKN1B) are transiently silenced to allow immunoglobulin affinity maturation2 (Figure 3). This process is critical for the formation and function of germinal center B cells that physiologically shut off BCL6 immediately after they exit the germinal center. Aberrant persistence of this status because of deregulated BCL6 expression through translocations or activating mutations promotes lymphomagenesis.43
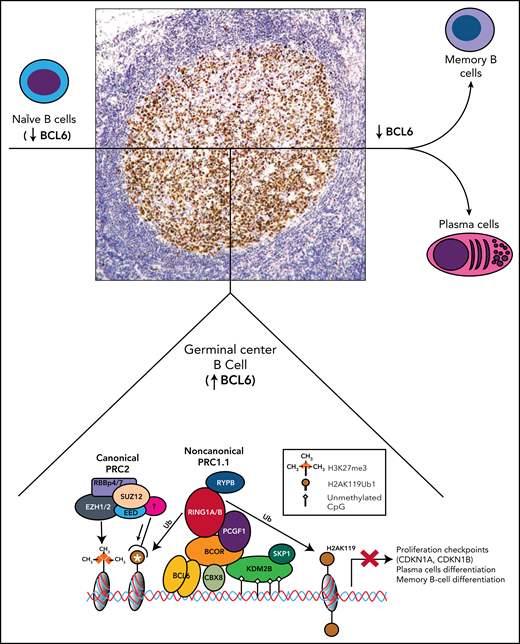
Role of BCOR and noncanonical PRC1.1 complex in the germinal centers of B-cell follicles. Mantle naïve B cells do not express BCL6. Germinal center B cells strongly express BCL6 (nuclear brown positivity at immunoperoxidase staining with monoclonal antibody PG-B6p42). BCL6 interacts with BCOR to recruits the PRC1.1 that leads to the epigenetic transcriptional repression of BCL6 target genes. CBX8 is also a component of the complex in the germinal center B cells.2 The white asterisk indicates recruitment of PRC2 to the ubiquitinated loci. In addition to BCOR, the POZ domain of BCL6 also interacts with the SMRT and N-CoR corepressors that are part of the large multiprotein histone deacetylase-containing complexes and are also required for the repressive activity of BCL6. These events result into the temporary silencing of genes controlling differentiation of B cells to plasma cells and cell cycle checkpoint (CDKN1A, CDKN1B) to allow immunoglobulin affinity maturation. B cells that exit from germinal center downregulate BCL6 before giving raise to plasma cells and memory B cells.
Role of BCOR and noncanonical PRC1.1 complex in the germinal centers of B-cell follicles. Mantle naïve B cells do not express BCL6. Germinal center B cells strongly express BCL6 (nuclear brown positivity at immunoperoxidase staining with monoclonal antibody PG-B6p42). BCL6 interacts with BCOR to recruits the PRC1.1 that leads to the epigenetic transcriptional repression of BCL6 target genes. CBX8 is also a component of the complex in the germinal center B cells.2 The white asterisk indicates recruitment of PRC2 to the ubiquitinated loci. In addition to BCOR, the POZ domain of BCL6 also interacts with the SMRT and N-CoR corepressors that are part of the large multiprotein histone deacetylase-containing complexes and are also required for the repressive activity of BCL6. These events result into the temporary silencing of genes controlling differentiation of B cells to plasma cells and cell cycle checkpoint (CDKN1A, CDKN1B) to allow immunoglobulin affinity maturation. B cells that exit from germinal center downregulate BCL6 before giving raise to plasma cells and memory B cells.
The Bcor-mediated recruitment of PRC1.1 complex by Bcl6 is also required for the differentiation of CD4+ T cells into follicular helper T cells (helping B cells to become plasma cells and memory cells), through repression of genes promoting differentiation toward other lineages.44,45 Independently by Bcl6, Bcor and Kdm2b in mice are both required to form CD4+ T helper 17 (Th17) cells that protect from extracellular pathogens at mucosal surfaces.46 Specifically, Bcor enhances Th17 development by repressing the Lef1, Runx2 (runt-related transcription factor 2), and Dusp4d (dual-specificity phosphatase 4) genes, encoding proteins that inhibit the Th17 cell fate.46
No information is currently available on the BCORL1 function. Association of BCORL1 hemizygous variants with the Shukla-Vernon syndrome suggests a potential role in neural development. Generation of a BCORL1 targeted mouse model is warranted to address this issue.
BCOR and BCORL1 gene alterations in human neoplasms
BCOR mutations mostly occur in hematologic malignancies and in mesenchymal tumors that curiously share histologic features (ie, small round blue cell appearance [Ewing-like sarcoma] or mixoid background and delicate capillary channels).47,48BCOR mutations are also detected in some central nervous system neoplasms and rare carcinomas.49
Unlike BCOR and BCORL1, other genes encoding components of the PRC1.1 complex are rarely mutated or deleted in hematologic malignancies. As far it concerns T cell malignancies, this may due to the fact that mice lacking the Pcgf1-binding domain of Bcor show a normal T lymphopoiesis,31 thereby providing more opportunities for transformation than mice insufficient for Kdm2b (another component of PRC1.1) that display severely impaired lymphopoiesis.40,50
In both myeloid and lymphoid malignancies BCOR mutations are scattered throughout the BCOR coding sequence, more frequently exon 4 (52.2%), and resemble germline BCOR mutations causing the OFCD syndrome11 (Figure 4A-B). The most common mutations are frameshifts (deletions and insertions; 36.83%) followed by nonsense and missense mutations (20.49% and 20.24%, respectively; supplemental Table 1; supplemental Figure 2). BCOR-mutated AML samples show low BCOR mRNA levels (mean of 22%), likely because of nonsense-mediated mRNA decay.9,51BCOR mutations result into the absence of full-length BCOR protein (192 kDa) and the lack or low expression of a truncated form of the protein of lower molecular weight.9 The disruptive nature of mutations is consistent with the tumor suppressor role of BCOR. BCORL1 mutations show similar features (supplemental Table 2; Figure 4C-D). Next-generation sequencing52 for copy number changes has also revealed BCOR deletions in AML.53
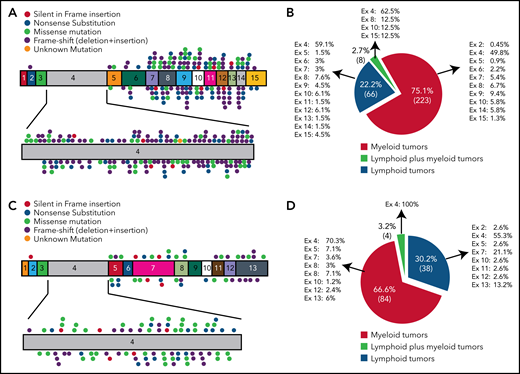
BCOR and BCORL1 mutations in hematologic malignancies. (A,C) The numbers indicate the coding exon, whereas the plots indicate the mutations in BCOR (A) and BCORL1 (C). Red plot = nonsense mutation; orange plot = silent in frame insertion; green plot = missense mutation; black plot = frameshift mutation (deletion + insertion); pink plot = unknown mutation. (B,D) Frequency of mutations for each BCOR (B) or BCORL1 (D) exon detected on myeloid neoplasms, lymphoid neoplasms, and in both of them.
BCOR and BCORL1 mutations in hematologic malignancies. (A,C) The numbers indicate the coding exon, whereas the plots indicate the mutations in BCOR (A) and BCORL1 (C). Red plot = nonsense mutation; orange plot = silent in frame insertion; green plot = missense mutation; black plot = frameshift mutation (deletion + insertion); pink plot = unknown mutation. (B,D) Frequency of mutations for each BCOR (B) or BCORL1 (D) exon detected on myeloid neoplasms, lymphoid neoplasms, and in both of them.
Mutations involving BCOR and other chromosome X genes (PHF6, STAG2, ZRSR2) were associated with male bias in AML.54 The fact that, in females, the mutation of BCOR (or PHF6) has no deleterious effect if it occurs in the X chromosome inactivated by lionization could explain the male predominance.54
Internal tandem duplications (ITDs) of PUFD domain do not occur in hematologic malignancies but are detected in kidney clear cell sarcoma and central nervous system high-grade neuroepithelial tumor with BCOR alteration (CNS-HGNET-BCOR), a rare pediatric aggressive brain tumor.55,56 Curiously, ITDs map to the structurally disordered BCOR PUFD termini, abrogating the binding to the PCGF1 RAWUL and preventing the formation of PRC1.1 complex.28 Notably, such alterations were not reported for BCORL1.28
Translocations involving BCOR are mainly detected in undifferentiated round cell sarcoma, high-grade endometrial sarcoma, and ossifying fibromyxoid tumor.47-49 Conversely, BCOR fusions were reported only in 2 patients with acute promyelocytic leukemia.57,58 In both cases, BCOR was translocated with the retinoic acid receptor α (RARα) gene because of a rare t(X;17)(p11;q12).57,58 Morphologically, 1 case showed Auer rods and Faggot cells, whereas the other did not. Both patients responded to all-transretinoic acid but experienced frequent relapses and were refractory to arsenic trioxide. The incidence (supplemental Table 1), significance, and clinical relevance of BCOR mutations in hematologicl diseases are discussed below.
BCOR and BCORL1 mutations in myeloid neoplasms
AML
BCOR mutations are detected in 3.8% to 5.0% of adult de novo AML9,59,60 and about 4% of AML with myelodysplasia-related changes.61 Frequency is lower in pediatric AML62 (BCOR 1.7%) and higher in secondary AML52,63 (about 8%). We found that about 45% of BCOR-mutated AML had concomitant DNMT3A and/or RUNX1 mutations and were mutually exclusive with FLT3 and NPM1 mutations.9 The hierarchy of BCOR, DNMT3A, and RUNX1 mutations in AML is poorly understood. BCOR-mutated AML patients also show a high rate of N-RAS and K-RAS mutations (36.8%).59BCORL1 mutations occur at a frequency of 3.7% to 5.8% of AML adult patients10,59 and 1.2% of pediatric AML patients.62 Coincidental BCOR and BCORL1 deleterious mutations were suggested to play a role (together with PHF6 mutation) in the leukemic transformation of a patient with familial platelet disorder related to germinal C-terminal RUNX1 mutation.64 Thus, occurrence of BCOR and BCORL1 mutations in the same tumor suggest the they may not be redundant, consistently with the likely different function of the 2 proteins.
Most BCOR-mutated AML patients show a normal karyotype.9 Cases with abnormal karyotype include the following: trisomy 8, t(9;11), −7, and complex karyotype.65 Trisomies 11 and 13, as well as inv(3)(q21q26)/t(3;3)(q21;q26), associate with high rate of BCOR mutations65-68 (25%-38%). In AML with t(16;21)(p11;q22)/FUS-ERG, BCOR mutations are frequent and appear to precede chromosomal translocation.69
BCOR-mutated AML shows a lower remission rate after induction (47.4%).59 In 422 de novo AML patients with normal cytogenetic, we found a shorter overall survival (OS) at 2 years in BCOR-mutated (25.6%) vs BCOR-unmutated (56.7%) patients.9 In a Japanese cohort of 377 de novo AML patients, BCOR mutations were associated with lower 5‐year OS and relapse-free survival, especially in patients ≤65 years of age, FLT3-ITD negative, and with intermediate cytogenetic prognosis.59 Similarly, in 509 Chinese patients, BCOR-mutated cases showed an inferior 2-year OS and 2-year relapse-free survival compared with unmutated cases.65 Hematopoietic stem cell transplantation seems to abrogate the adverse prognostic impact of BCOR mutations.59,65
The 2017 EuropeanLeukemiaNet risk stratification does not regard BCOR mutations as a prognostic predictor,70 but their inclusion in the intermediate risk group was recently proposed.71 Mutations of BCOR, other epigenetic modifiers, and RNA-splicing regulators also define a heterogeneous category (18% of AML) with intermediate/adverse prognosis of the genetic classification of AML.60BCOR mutations predicted complete response to venetoclax plus hypomethylating agents in AML.72,73
Myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms
BCOR mutations occur in 4.2% to 5.0% of myelodysplastic syndromes (MDS) (vs 0.8%-2.0% of BCORL1 mutations),51,74 who usually carry a normal karyotype75 and are comutated for RUNX1 and DNMT3A.51,74,76,77BCOR mutations also associate with mutations of ASXL1, NF1, ETV6, BCORL1, MECOM, RAD21, CEBPA, and Cohesin genes.74,78BCOR mutations occur in all International Prognostic Scoring System (IPSS) risk groups and World Health Organization subtypes,74 being more frequent in patients younger and with lower platelet counts at diagnosis.51,74 Cryptic recurrent deletions at Xp11.4 (where BCOR is located) are detected in 2.8% MDS patients with normal or noninformative karyotype.79
The BCOR mutant-copy burden in flow-sorted CD34+/CD38− early hematopoietic progenitors is lower than that of RUNX1, STAG2, and ASXL1 mutations,51 suggesting that BCOR mutations hierarchically occur at a later stage and define the clinical course rather than initiation of MDS.51
The prognostic impact of BCOR mutations in MDS remains controversial.51,74,76 Two studies on about 1000 patients showed inferior OS.51,76 In another cohort of 621 patients, BCOR and BCORL1 mutations did not impact OS, whereas the mutation type did. In particular, patients carrying frameshift mutations showed a median OS74 lower than those with other types of mutations.74 Moreover, mutations at the C terminus of BCORL1 were associated with an OS shorter than mutations outside the C terminus.74 However, these findings need to be confirmed.
Finally, MDS patients with isolated BCOR mutations showed a trend toward a prognosis poorer than cases comutated for BCOR and TET2, ASXL1, or DNMT3A.80 This may be due to enrichment of poor cytogenetic in the BCOR-mutated-only group or to better response to hypomethylating agents when other epigenetic modulators are comutated.80
BCOR mutations occur in 3% to 10% of chronic myelomonocytic leukemia (CMML),35,81,82 especially CMML-2, which is frequently comutated for U2AF1 and RUNX151 or ASXL1/EZH2.83 The prognostic value of BCOR mutations in CMML remains uncertain.84,85BCOR mutations also occur in 24% of MDS/myeloproliferative neoplasms with ring sideroblasts and thrombocytosis.82,86 Notably, increased median corpuscular volume of erythrocytes and thrombocytosis was observed in our conditional knockout Bcor mice.87
Myeloproliferative neoplasms
BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia (CML).88BCOR mutations are rare in chronic-phase CML and, when present, usually persist despite marked reduction of BCR-ABL transcript following tyrosine kinase inhibitors. Thus, they may originate from a preleukemic Philadelphia-negative (Ph−) clone that existed independently of Ph1+ clones.89BCOR mutations occur in about 16% of blastic-phase CML90 and contribute driving CML transformation.91BCOR and ASXL1 mutations also appear to be independent predictors for worse response to tyrosine kinase inhibitors of blastic-phase CML.92
A missense BCOR mutation was reported in 1 essential thrombocytemia patient triple negative for JAK2, CALR, and MPL and with a normal karyotype,93 presenting with a high platelet count and resistant to therapy. Curiously, we observed thrombocytosis in our conditional knockout Bcor mice.87BCORL1 and RUNX1 mutations occurring concomitantly with a novel mutation of JAK2 at serine 523 were detected in a patient with increased hematocrit.94 Moreover, mutations of BCORL1 (together with TP53 and NRAS) have been associated with long-term (>21 years) leukemic transformation of polycythemia vera and essential thromcocytopenia.95 However, this event is very rare.96
BCOR mutations in B-cell malignancies
The somatic mutation rate of B-cell chronic lymphocytic leukemia is low, including up to 2% of BCOR mutations.97,98 Despite the low incidence of B-cell chronic lymphocytic leukemia in Asia, BCOR mutations tend to be more frequent in Korean than White patients.99 Most BCOR-mutated cases are IGHV unmutated,100 carry trisomy 12, and are NOTCH1 mutated.101,102
B-cell prolymphocytic leukemia (B-PLL) usually carries a complex karyotype with frequent MYC translocations or gains and (del)17p.103 About 25% of B-PLL patients harbor BCOR mutations103 that are usually early clonal events.103BCOR mutations likely cooperate with MYC translocations in promoting B-PLL. BCOR mutations were detected in 9% of mantle cell lymphoma patients104 and may cooperate with KDM5C mutations by increasing H3K4 and try-methylation in the late stages of mantle cell lymphoma.105
Clonal BCOR mutations or losses occurred in 24% of splenic diffuse red-pulp small B-cell lymphoma,106 whereas they were absent in hairy cell leukemia and hairy cell leukemia variant and were only rarely found in splenic marginal zone lymphoma. Other B-cell lymphomas mutated for BCOR are listed in supplemental Table 1.
BCOR mutations in T-cell malignancies
BCOR mutations were detected in 2% to 3% of pediatric T-cell acute lymphoblastic leukemia with high TAL1 expression, being mutually exclusive with TLX1 and TLX3 expression (TLX-related).107
T-cell prolymphocytic leukemia is an aggressive disease carrying the inv(14) (q11q32)/t(14;14) (q11;q32) or t(X;14)(q28;q11). However, other genetic alterations may contribute to promote T-cell prolymphocytic leukemia, such as BCOR mutations that occur in 8% to 9% of cases.108,109BCOR deletion, at Xp11.4, is also revealed by Comparative Genomic Hybridization (CGH) array.108
Extranodal natural killer/T-cell lymphoma nasal type (ENKTL) frequently carries mutations in the JAK‐STAT pathway.110BCOR mutations occur in 20.6% to 32% patients,110,111 suggesting they may play a pathogenetic role in ENKTL.112 However, the BCOR K607E mutation is not restricted to natural killer/T-cell lymphomas (31.9%), being also observed in angioimmunoblastic T-cell lymphomas (11.1%) and peripheral T-cell lymphomas not otherwise specified (33.3%).113 Because Epstein-Barr virus infection promotes ENKTL through epigenetic mechanisms,114BCOR mutations could cooperate with Epstein-Barr virus by amplifying epigenetic deregulation.
BCOR and BCORL1 mutations in nonmalignant hematologic diseases
Acquired aplastic anemia
Aplastic anemia (AA) shows a high frequency of clonal hematopoiesis.115,116 Mutations of BCOR, BCORL1, DNMT3A, ASXL1, and PIGA have been detected in AA.117-120 Frequency of BCOR/BCORL1 mutations in AA ranged between 0% and 10.9%.118-122 This suggests that, when such mutations occur without a proper ancestral hit, they are possibly unable to drive an efficient clonal expansion and are thus overridden once normal polyclonal hematopoietic stem cells recover.
AA patients carry a disproportionate number of BCOR and BCORL1 mutations compared with their expected frequency in an age-matched population. Thus, these mutations are more likely to be selected by the AA bone marrow milieu rather than representing an age-related outgrowth.120,122 In AA, the autoimmune attack of T lymphocytes against HSCs can result into selective growth advantage of cells that, by acquiring somatic mutations, become less immunogenic.117 Unlike DNMT3A and ASXL1 mutations, those involving PIGA, BCOR, and BCORL1 tended to disappear or showed stable clone size. Moreover, AA patients carrying BCOR, BCORL1, and PIGA mutations responded better to immunosuppressive therapy than patients with other mutations and showed a good OS and progression-free survival.120 Conversely, DNMT3A and ASXL1 mutations tended to increase their clone size and were associated with worse outcome.120 Similar findings have been reported for BCOR and BCORL1 mutations in pure red cell aplasia.123
MDS and AML usually develop in 15% to 26% of AA patients over a period of 10 years.115 Unlike high-risk ASXL1 and RUNX1 mutations that promote evolution of AA to MDS/AML, BCOR and BCORL1 mutations impart a low risk of transformation into MDS/AML.115,124,125
Erythrocytosis
Erythrocytosis defined by the strict 2008 World Health Organization classification criteria (hemoglobin > 18.5 g/dL or Hematocrit (Hct) ≥ 52% in males; hemoglobin > 16.5 g/dL or Hct ≥ 48% in females), associates with cardiovascular morbidity/mortality and all-cause mortality,126 independently of conventional risk factors. Moreover, cardiovascular morbidity is strongly associated with clonal hematopoiesis, mostly because of BCOR/BCORL1 mutations (16%).126 Similar to AA, BCOR/BCORL1 mutations in erythrocytosis associate with a low risk of transformation into MDS/AML.126
Role of BCOR in the pathogenesis of hematologic malignancies
Given the disruptive nature of BCOR mutations, the role of BCOR in promoting hematologic malignancies was mainly investigated in animal models whose endogenous gene had been inactivated. Initial Bcor loss-of-function studies in zebrafish and Xenopus recapitulated the phenotype of OFCD syndrome.127Bcor knockout mouse models are discussed below.
Myeloid malignancies
A conditional loss-of-function model targeting exons 9 and 10 of the Bcor allele allowing their removal via expression of either a retrovirus-expressing Cre ex vivo or a Vav-iCre recombinase in vivo was generated.37 Excision led to a premature stop codon with deletion of carboxy-terminal Bcor domain required for proper formation of PRC1.1 complex.24Bcor mutant cells cultured under myeloid stem/progenitor conditions showed higher proliferation rates than control cells. Bcor knockout mice exhibited a marked increase of peripheral blood neutrophils without significant changes in red blood cells, platelet and lymphocyte levels. Conversely, other investigators failed to demonstrate peripheral blood counts alterations, using the same conditional model crossed with HSC-SCL-Cre-ERT mice to facilitate tamoxifen-inducible Bcor deletion specifically in HSCs.128 Nevertheless, mutant mice showed expanded BM cKit+Sca1−Lin− myeloid progenitors with enhanced repopulating capacity in vivo.128 Both mouse models exhibited overexpression of Hox genes. Specifically, Bcor loss reduced the levels of RING1B in the complex, leading to a reduced monoubiquitylation of H2A at position 119 of HoxA promoters with consequent upregulated Hox transcription.
More recently, we developed a conditional Bcor knockout mutant87 targeting exons 8 to 10 resulting in a premature stop codon in exon 11 and tested the effects of Bcor loss in hematopoiesis using Mx1-Cre mice.87 Mice displayed leukopenia, mainly because of B-cell lymphopenia, red blood cell reduction with increased mean corpuscle volume, and progressive increase of platelet counts. Thrombocytosis was caused by accumulation of megakaryocytic-erythroid and megakaryocytic progenitors due to apoptosis resistance. Thus, Bcor loss of function induces derepression of Hox and Cebp family genes,37 myeloid differentiation, and thrombocytosis.87 However, it is insufficient to promote myeloid malignancies alone, clearly pointing to the need of additional cooperative events, as depicted in Figure 5.
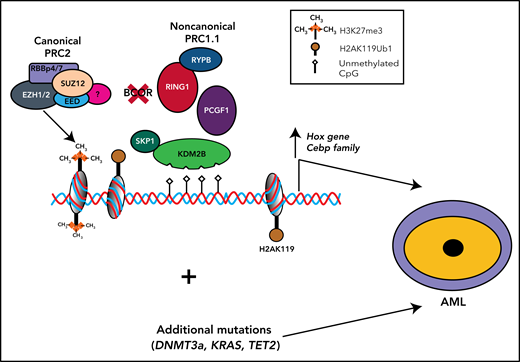
BCOR functional loss cooperates with other mutations to promote AML. Disruptive BCOR mutations that cause loss of the native protein or generate a truncated protein abrogate the capability of BCOR to bind PCGF1, thus preventing its interaction with KDM2B and the formation and recruitment to chromating of the enzymatic core. Thus, in hematopoietic stem and progenitor cells (HSPCs), the repressive activity of the complex is abrogated resulting into the expression of Hox and Cebp family genes. The occurrence of additional mutations (eg, DNMT3A and RUNX1) promotes the development of AML. Whether BCOR mutations precede or follow DNMT3A and RUNX1 mutations remains to be established.
BCOR functional loss cooperates with other mutations to promote AML. Disruptive BCOR mutations that cause loss of the native protein or generate a truncated protein abrogate the capability of BCOR to bind PCGF1, thus preventing its interaction with KDM2B and the formation and recruitment to chromating of the enzymatic core. Thus, in hematopoietic stem and progenitor cells (HSPCs), the repressive activity of the complex is abrogated resulting into the expression of Hox and Cebp family genes. The occurrence of additional mutations (eg, DNMT3A and RUNX1) promotes the development of AML. Whether BCOR mutations precede or follow DNMT3A and RUNX1 mutations remains to be established.
Because BCOR and DNMT3A are frequently comutated in AML,9 we generated Bcor/Dnmt3a double knockout mice87 that rapidly developed a lethal leukemic phenotype characterized by immature erythroid cells expansion87 (Figure 6). The aberrant erythroid skewing was induced by an altered molecular program affecting major cell cycle regulators (Mdm2, Tp53) and erythroid-specific transcriptional factors (Gata1-2)87 (Figure 6). Another mouse model of acute erythroid leukemia involving loss of Bcor and Dnmt3a (in addition to Trp53) and characterized by deregulation of aberrantly methylated driver genes has been recently reported129 (Figure 6).

Schematic representation of BCOR knockout and double compound mouse models. The presence of different partner mutations in the Bcor conditional knockout mouse model (BcorΔ4/Y, BcorΔ9-10/Y, BcorΔ8-10/Y) variably affects the severity and penetrance of the disease phenotype. In particular, BcorΔ4/Y,p53−/− mice exacerbate the T-ALL developed in BcorΔ4/Y mice. Compound mutant mice carrying BcorΔ9-10/Y,Tet2−/− mutations develop a progressive lethal MDS. Compound mice comutated for BcorΔ9-10/Y, and KrasG12D develop AML, and mice comutated for BcorΔ8-10/Y and Dnmt3aΔ19-20 develop acute erythroid leukemia (AEL). An approach of multiplex genome editing of primary mouse hematopoietic stem and progenitor cell transplanted in a clustered regularly interspaced short palindromic repeat (CRISPR)-cas9 mice compound demonstrates that comutations of Bcor, Trp53 plus Dnmt3a or Rb1 or Nfix results in AEL. In contrast, the contemporary comutations of Bcor, Dnmt3a, Trp53, and tet2 result in T-ALL, and the contemporary comutations of Bcor, tet2 and Sf3b3 lead to B-ALL.
Schematic representation of BCOR knockout and double compound mouse models. The presence of different partner mutations in the Bcor conditional knockout mouse model (BcorΔ4/Y, BcorΔ9-10/Y, BcorΔ8-10/Y) variably affects the severity and penetrance of the disease phenotype. In particular, BcorΔ4/Y,p53−/− mice exacerbate the T-ALL developed in BcorΔ4/Y mice. Compound mutant mice carrying BcorΔ9-10/Y,Tet2−/− mutations develop a progressive lethal MDS. Compound mice comutated for BcorΔ9-10/Y, and KrasG12D develop AML, and mice comutated for BcorΔ8-10/Y and Dnmt3aΔ19-20 develop acute erythroid leukemia (AEL). An approach of multiplex genome editing of primary mouse hematopoietic stem and progenitor cell transplanted in a clustered regularly interspaced short palindromic repeat (CRISPR)-cas9 mice compound demonstrates that comutations of Bcor, Trp53 plus Dnmt3a or Rb1 or Nfix results in AEL. In contrast, the contemporary comutations of Bcor, Dnmt3a, Trp53, and tet2 result in T-ALL, and the contemporary comutations of Bcor, tet2 and Sf3b3 lead to B-ALL.
BCOR and RAS mutations cooccur in both AML and MDS. Bcor-deficient mice crossed with Kras mutant animals128 developed a lethal disease characterized by leucocytosis, splenomegaly, and increased leukemic blasts through Hoxa 9 upregulation (Figure 6). BCOR and TET2 are also frequently comutated in MDS patients.51,76,130 Accordingly, Bcor and Tet2 disruption in mice induced a lethal MDS phenotype with differentiation block, apoptosis, and activation of myeloid regulator genes of the Cebp and Hoxa family through reduction of H2AK119ub levels31 (Figure 6).
One of the BCOR functional domains (Figure 1) directly binds to the common MLL fusion partner AF9 (MLLT3), contributing to promote MLL rearranged leukemia.131 Mutagenesis studies identified point mutations selectively disrupting the capability of BCOR to bind MLLT3. Expression of these BCOR point mutations in BM stem/progenitor cells caused partial differentiation and abrogated the leukemogenic potential in a mouse model131 through downregulation of EYA1 phosphatase and c-MYC protein expression.131
We recently found BCOR mutations in the AML cell lines MUTZ-2, KG-1, and HL60-R (E. Tiachi and B. Falini, unpublished data). BCOR reconstitution in HL60-R cells inhibited cell growth and increased vitamin D3–induced differentiation (E. Tiachi and B. Falini, unpublished data). BCORL1 mutations were detected in the AML-193, SKM1, and OCI-AML5 cell lines.10 The MUTZ-2 AML cell line carries both BCOR and BCORL1 mutations.10 All these cell lines may serve for functional studies and drug testing in vitro and in vivo.
Lymphoid malignancies
About 10% of NUP98-PHF2 (NP23) transgenic mice develop an aggressive pro-B1 ALL that carries spontaneous Bcor indel mutations, leading to premature stop codons, usually within a 9-bp hotspot in exon 8.132,133 Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 insertion of Bcor frameshift mutation into NP23 hematopoietic stem/progenitor cells and their transplantation into recipient irradiated mice led to pro-B1 ALL development,134 suggesting a cooperation between mutated Bcor and NP23 fusion. The human counterpart of mouse B-1 cells remains elusive, and Bcor mutations are very rare in human B-cell ALL. However, mouse NP23/Bcor pro-B1 ALL tends to acquire Jak mutations and may serve as a model for human B-progenitor ALL with Jak mutation and rearrangements causing overexpression of CRLF2,132 a receptor for thymic stromal lymphopoietin critical for B-1 cell development.
Emu-Myc mice spontaneously develop a B-cell leukemia/lymphoma–like malignancy with 100% penetrance. Destructive Bcor mutations and loss of Cdkn2a cooperate with overexpressed Myc to promote this disease.135
Mice expressing Bcor lacking the BCL6-binding domain136 showed impaired B lymphopoiesis, and 50% of animals developed lethal T-ALL with late latency. However, the concomitant p53 loss accelerated T-ALL development (Figure 6). Thymic leukemic blasts displayed activated Notch1 and upregulation of its target genes Myc (via Bcor loss of function135) and Hes1. These findings suggest a tumor suppressor role for Bcor in T-ALL, antagonizing the transcriptional activation of T-ALL related oncogenes by Notch1.136Bcor loss of function may induce leukemia abrogating the PRC1.1 complex formation because mice without the ZF-CxxC DNA-binding domain of Kdm2b develop T-ALL.50
BCOR carrying the K607E mutation (located near the BCL6 binding site) binds to BCL6, PCGF1, and RING1B proteins with lower affinity than BCOR wild type.113 Ectopic expression of BCOR-K607E mutant drives the constitutive activation of T cells (ie, enhanced cell proliferation, increased phosphorylation of AKT, and production of interleukin-2). Similar effects were mimicked by silencing BCOR in T cells.113 Similarly to AML, the BCOR mutant led to upregulation of HOX genes.113
Conclusions and perspectives
BCOR is involved in the regulation of embryogenesis, mesenchymal stem cell function, hematopoiesis, and lymphoid development. BCOR and BCORL1 disruptive mutations contribute to the origin of various hematologic malignancies and are similar to those found in OFCD and Shukla-Vernon syndromes.
Therapeutic targeting of BCOR-containing PRC1.1 complex functions (eg, H2AK119 ubiquitylation and H3K36 demethylation) should be explored.137 AML cells with BCOR mutations alone are sensitive to the tankyrase/WNT inhibitor XAV-939 and the multikinase inhibitor crizotinib.138BCOR/RUNX1-comutated AML cells are sensitive to JAK kinase inhibitors,138 whereas acute erythroid leukemia driven by Bcor and Dnmt3a loss is susceptible to CDK7/CDK9 inhibitors.129 Better understanding of the role played by BCOR and BCORL1 in leukemogenesis and screening for synthetically lethal partners of these mutations may help unraveling new therapeutic opportunities.
Acknowledgments
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC; IG 2019 23604) and the ARC Foundation for Cancer Research (Leopold Griffuel Prize [B.F.]).
The authors apologize to those whose papers could not be cited because of the space limitation.
Authorship
Contribution: P.S., D.S., and B.F. conceived the study and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Brunangelo Falini, Institute of Hematology and Center for Hemato-Oncological Research (CREO), University of Perugia, Piazzale Menghini 8/9, Perugia, 06132, Italy; e-mail: brunangelo.falini@unipg.it.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal