

Key Points
A murine model of anti-CD36–mediated FNAIT showed frequent fetal death in immunized mothers that could be prevented by IVIG and deg-mAb.
Therapy with deg-mAb against CD36 is more beneficial than IVIG based on the dose, the success rate of therapy, and the start of application.

Abstract
Recent studies have shown that maternal anti-CD36 antibodies represent a frequent cause of fetal/neonatal alloimmune thrombocytopenia (FNAIT) in Asian and African populations. However, little is known about the pathomechanism and antenatal treatment of anti-CD36–mediated FNAIT. Here, we established a novel animal model to examine the clinical features of pups from immunized Cd36−/− female mice after breeding with wild-type male mice. Mild thrombocytopenia was observed, but high pup mortality was also documented (40.26%). Administration of intravenous immunoglobulin (IVIG) (1 g/kg) on days 7, 12, and 17 to immunized Cd36−/− mothers after breeding reduced fetal death (12.70%). However, delaying the IVIG administration series on days 10, 15, and 20 did not reduce fetal death (40.00%). In contrast, injection of deglycosylated anti-CD36 (deg-anti-CD36) polyclonal antibodies (5 mg/kg) on days 10, 15, and 20 significantly reduced fetal death (5.26%). Subsequently, monoclonal antibodies (mAbs) against mouse CD36 were developed, and one clone producing high-affinity anti-CD36 (termed 32-106) effectively inhibited maternal antibody binding and was therefore selected. Using the same approach of deg-anti-CD36, the administration of deg-32-106 significantly reduced fetal death (2.17%). Furthermore, immunized Cd36−/− mothers exhibited placental deficiency. Accordingly, maternal anti-CD36 antibodies inhibited angiogenesis of placenta endothelial cells, which could be restored by deg-32-106. In summary, maternal anti-CD36 antibodies caused a high frequency of fetal death in our animal model, associated with placental dysfunction. This deleterious effect could be diminished by the antenatal administration of IVIG and deg-mAb 32-106. Interestingly, treatment with deg-32-106 seems more beneficial considering the lower dose, later start of treatment, and therapy success.
Introduction
Fetal and neonatal alloimmune thrombocytopenia (FNAIT) is caused by maternal antibodies that cross through the placenta via neonatal Fc receptor (FcRn)-mediated transport during pregnancy. This action can lead to the clearance of fetal platelets and endothelial dysfunction, which results in bleeding complications.1-3 In White populations, antibodies against human platelet antigen-1a (HPA-1a) are responsible for ∼80% of FNAIT cases.4 The incidence of severe FNAIT is ∼1:2500 newborns,5 in which intracranial hemorrhage (ICH) occurs in ∼20% of these cases, leading to fetal death or persistent neurologic sequelae.6 However, FNAIT caused by anti–HPA-1a antibodies has not been well recognized in other populations.
The data indicate that antibodies against CD36 represent an important risk factor for FNAIT in Asian and African populations.7-10 CD36 is a receptor for thrombospondin-1, a free fatty acid, and a scavenger receptor for different danger signals. Accordingly, CD36 is implicated in inflammatory response, atherosclerosis, thrombosis, and metabolic disorders.11 Individuals lacking CD36 on both platelets and monocytes (type I) are at risk of developing anti-CD36 antibodies (known as anti-Naka) after receiving a platelet transfusion or during pregnancy.12,13 These antibodies may cause immune-mediated thrombocytopenia and repeated early fetal loss.14-16 Immunization in type II individuals (lacking CD36 on platelets only but not on monocytes) has not been described. Type I CD36 deficiency is extremely rare in White subjects but more common in African subjects (∼2%) and relatively frequent in Asian populations (>0.5%).7,9,17-19
The clinical manifestation of FNAIT caused by anti-CD36 antibodies is heterogeneous, ranging from widespread petechial hemorrhages to thrombocytopenia, miscarriages, hydrops fetalis, and ICH. Compared with anti–HPA-1a, anti-CD36 antibodies cause more common hydrops and recurrent miscarriages.16,20,21 The reason for this phenomenon is unknown. Therefore, the current approach for treating FNAIT caused by anti–HPA-1a may not be adaptable for the disease caused by anti-CD36 antibodies.
The current clinical approach for managing FNAIT relies on preventing fetal bleeding complications through antenatal therapy because the prophylaxis is currently unavailable.22 Presently, noninvasive maternal treatment with intravenous immunoglobulin (IVIG) represents the antenatal therapy’s first line.1 However, such strategies may inhibit the transfer of immune-protective maternal immunoglobulin G (IgG), which might cause an increased risk of infection during pregnancy and the first weeks following birth.23 An attractive alternative strategy would be to administer nondestructive IgG that shares the pathogenic antibodies’ specificity and retains the ability to be transported across the placenta. Ghevaert et al24,25 introduced such a strategy using human recombinant single-chain fragment variable anti–HPA-1a antibodies with a modified Fc part. These modified recombinant anti–HPA-1a antibodies could pass the placenta and ease FNAIT in immunized mothers.
The therapeutic potential of deglycosylated IgG (deg-IgG) antibodies for autoimmunity treatment has been widely recognized.26 Removal of N-glycan (linked to asparagines 297), located on the Fc part, leads to a significant reduction of IgG binding with the FcγRs expressed on macrophages and its ability to activate complement C1q. Interestingly, these deg-IgG antibodies could still be transported from the maternal circulation to the fetus via FcRn.27 Accordingly, our previous in vivo study in mice showed that deglycosylated–monoclonal antibody (deg-mAb) specific for HPA-1a could pass through the placenta and prevent the clearance of fetal platelets mediated by maternal anti–HPA-1a antibodies. This observation indicates that the use of epitope-specific antibodies for the antenatal therapy of severe FNAIT is feasible.28
The current study established a novel mouse FNAIT model to evaluate the efficacy of deglycosylated anti-CD36 (deg-anti-CD36) to treat FNAIT caused by maternal anti-CD36 antibodies and compared this treatment with IVIG.
Materials and methods
Intravenous immunoglobulin (IVIG)
Human IVIG was purchased from Jiangxi Boya Bio-Pharmaceutical, Jiangxi, China.
Mice
Wild-type (WT) C57BL/6J mice were provided by the Animal Centre of Sun Yat-Sen University, China. Cd36−/− mice were purchased from The Jackson Laboratory (B6.129S1-Cd36tm1Mfe/J). The Animal Care Committee approved this study (IACUC-2014-0303).
Immunization of Cd36−/− mice and detection of anti-CD36 antibodies
Platelets were prepared as previously described.29Cd36−/− female mice (6-8 weeks old) were immunized 3 times with 108 WT platelets intraperitoneally at weekly intervals. After immunization, 5 µL of serum was collected and incubated with 100 µL of EDTA blood (1:100) from WT mice for 30 minutes and washed with phosphate-buffered saline/1% bovine serum albumin. Then, 50 µL of fluorescein isothiocyanate–conjugated anti-mouse IgG (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA) was added for 30 minutes. After red blood cell lysis (BD Biosciences, Shanghai, China), cells were suspended in 0.5 mL phosphate-buffered saline/1% bovine serum albumin and then analyzed by flow cytometry (FACS Canto II; BD Biosciences).
Counting of pups’ platelets
Platelets from the pups were counted as previously described.29 Briefly, 50 µL of counting beads (Life Technologies, Carlsbad, CA) was added to determine the platelet counts by using flow cytometry. Counting of 2,000 beads represented the standard.
Generation and characterization of mAbs against CD36
Cd36−/− mice were immunized as previously described.3,29 Splenocytes were harvested and fused with SP2/0-Ag14 mouse myeloma cells (ATCC, Manassas, VA) with the help of polyethylene glycol using a standard protocol.30 Hybridomas were screened by using mouse platelets according to flow cytometry. IgG subclasses were determined by a Pierce Rapid Isotyping Kit (Thermo Fisher Scientific, Waltham, MA). Seventeen hybridoma clones producing anti-CD36 could be generated. One clone (labeled 32-106) producing a high-affinity IgG antibody that effectively blocked maternal anti-CD36 binding was selected.
Deglycosylation of anti-CD36 antibodies
IgG was purified and digested as previously described.28 Aliquots of 1 µL of native and deg-mAb (0.6 mg/mL) were analyzed by flow cytometry using mouse IgG2a as isotype control (eBioscience, San Diego, CA).
Competitive inhibition between maternal and mAb against CD36 in vitro
One milligram of IgG derived from immunized Cd36−/− mice was labeled with fluorescein-EX (Invitrogen, Carlsbad, CA). Unlabeled deg-32-106 (5-50 ng) was added to 100 µL of EDTA- anticoagulated mice blood at room temperature for 30 minutes. After washing, 8 µL of fluorescein-conjugated IgG (1.73 mg/mL) was added for 30 minutes. Red blood cells were lysed and the samples were analyzed by flow cytometry.
Competitive inhibition between maternal and mAb against CD36 in vivo
Deg-32-106 (100 µg) was labeled with Alexa Fluor dye (AF-647; Invitrogen). Subsequently, Alexa Fluor–labeled deg-32-106 was intravenously injected into WT female mice (1 mg/kg). After 10 minutes, fluorescein-EX conjugated maternal IgG containing anti-CD36 was administered (200 mg/kg). The binding of both antibodies on platelets was analyzed by flow cytometry at 30 and 60 minutes. A single administration of labeled deg-32-106 or maternal anti-CD36 was run as a control.
Induction of FNAIT and treatment with IVIG or deg-anti-CD36 antibodies
Naive or immunized Cd36−/− female mice were bred with WT male mice. Anti-CD36 developed in Cd36−/− mothers was monitored by flow cytometry. During pregnancy, mothers were untreated or treated with human IVIG (1 g/kg), deg-anti-CD36 polyclonal IgG (5 mg/kg), or deg-anti-CD36 mAb (5 mg/kg) intravenously 3 times.3,29 Dead pups were determined in utero or within 24 hours after delivery. Miscarriage during pregnancy (a drop of body weight >1 g), platelet counts (within 24 hours), and bleeding in the pups after delivery were analyzed, as previously described.31
Histologic analysis of murine placenta
Murine placentas were collected at 16.5 days’ postcoitum and embedded in paraffin. Placental sections (3 µm) were stained with hematoxylin/eosin to determine the labyrinth zone ratio of the whole placenta. For immunohistochemistry, placental sections were incubated with anti-CD31 at 4°C overnight and stained with enzyme-labeled secondary antibody (Servicebio, Wuhan, China). Two fields per placenta labyrinth zone were selected, and the CD31+ area was quantified by using Image J (National Institutes of Health, Bethesda, MD).
Tube formation assay
Human placental microvascular endothelial cells (HPVEC; 4 × 105/mL; FuDan, IBS Cell Center, Shanghai, China) in 50 µL Dulbecco’s modified Eagle medium (serum free) were seeded onto the Matrigel (Corning, Bedford) and incubated for 30 minutes at 37°C. Thrombin (1 U; MilliporeSigma, Burlington, MA), 4 µg mouse IgG, mAb 32-106, or deg-32-106 in 50 µL Dulbecco’s modified Eagle medium containing 5% mouse serum was added. For the blocking experiment, 1 µg anti-FcγRIIa (clone IV.3; Stemcell Technologies, Vancouver, BC, Canada) was added with mAb 32-106. Furthermore, 50 µL anti-CD36 sera (1:10, 1:40) was transferred into the gel and incubated with HPVEC at 37°C for 6 hours in the absence or presence of deg-32-106. Images were taken from 2 to 3 selected areas per well. The tube formation assay was repeated 3 times. The total tube length was quantified by using Wimasis Image Analysis (Onimagin Technologies, Córdoba, Spain).
Statistical analysis
Data are presented as mean ± standard deviation and were analyzed by using Prism version 5.0 (GraphPad Software, La Jolla, CA). Comparisons of the 2 groups were assessed by using the two-tailed unpaired Student t test and χ2 test. One-way analysis of variance was used for the multiple comparisons, and the Fisher exact test was used for small sample sizes. Correlations between anti-CD36 titers and mortality rates were analyzed by using the Pearson correlation method. P values <.05 were considered significant.
Results
Antibodies developed in immunized Cd36−/− female mice caused severe FNAIT in the animal model
We initially investigated whether Cd36−/− female mice could develop natural anti-CD36 antibodies during the first pregnancy when crossed with WT male mice. An analysis of maternal sera by flow cytometry showed that immunization rarely occurred. Only one-fifth of mothers developed anti-CD36 at low titers and only after the second and third pregnancy. Interestingly, one mother (#1) delivered a smaller littermate (litter size, 1.50 ± 0.71) compared with mothers (#2-5) that lacked anti-CD36 antibodies (litter size, 7.50 ± 1.93) (Figure 1A), indicating that miscarriages and fetal death occurred during pregnancy.
![The effect of maternal anti-CD36 antibodies on the fate of pups delivered by immunized Cd36−/− female mice. (A) Naive Cd36−/− female mice (n = 5) were bred with WT male mice and delivered pups 3 times. The litter size was documented. One mother (#1) delivered a smaller littermate after the second and third pregnancy (litter size, 1.50 ± 0.71) compared with the other 4 mothers (#2-5) (litter size, 7.50 ± 1.93). Weak CD36-reactive antibodies were detected in mother #1 according to flow cytometry (see inset, the third delivery) but not in mothers #2 to #5 (data not shown). (B) Cd36−/− female mice (cohort #3) were immunized 3 times with WT platelets. Representative sera were then incubated with platelets from WT (white curves) or Cd36−/− mice (gray curves). Bound antibodies were detected with fluorescence-labeled anti-mouse IgG and analyzed by using flow cytometry. The percentages of positive cells (anti-CD36) are given. Sera from naive Cd36−/− were used as controls. (C) After immunization, Cd36−/− female mice (cohort #3; n = 16) were bred with WT male mice. Naive WT (cohort #1; n = 5) and naive Cd36−/− (cohort #2; n = 4) were conducted as controls. Dead pups were determined in utero or within 24 hours after delivery. The mortality of the pups in cohort #3 (31 of 77 [40.26%]) was compared with that of cohort #1 (χ2 = 17.46; ***P < .0001) and cohort #2 (χ2 = 11.52; ***P = .0007). (D) Severe bleeding in Cd36+/− pups delivered by immunized Cd36−/− mothers was found. (a) Healthy pup; (b) dead pup with bleeding (arrow); (c) dead pup with ICH (arrow); (d) dead pup with hydrops. (E) The litter size (including the dead pups) in naive WT, Cd36−/− mothers (cohorts #1 and #2), and immunized Cd36−/− mothers (cohort #3) is presented. The litter size in immunized Cd36−/− (cohort #3) was small but not significantly different compared with the naive WT (cohort #1) (4.81 ± 2.69 vs 7.40 ± 2.07; P = .064) and Cd36−/− (cohort #2) mothers (4.81 ± 2.69 vs 6.25 ± 1.50; P = .323) using a two-tailed unpaired Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/18/10.1182_blood.2021011131/4/m_bloodbld2021011131f1.png?Expires=1770127970&Signature=sFHhGx2AxI2SGA2eAyxELTt1BjICkv0xCTqOQ0rRwMu6o7tHtKz3UDTpVF4jXvnFPBoE8CaBnbzUyo7WQ9hVP4cNDXIKin-zUwb3kWfXi8A7YXsEEeAX5wmKYCV9orqgrLzTVijKigZ8zoadALNxOcqF9e-XcYpAEbvRviX~lavgabgvYQ1vwTIs7ju5ppQpLZrRXif9itYECLcGc8YMuRV1FM-HPurRBrM9q2Qqoa2jodxEdOvQ5e-0HQsqXliugOdBgd~YzgVJQusz7zNjNLHKmFSRMLV0Pf5sSSWKBgFeo5j0AAn29Ky5wJ~D15r3IJMdJGobohn56I0Qen0fqw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The effect of maternal anti-CD36 antibodies on the fate of pups delivered by immunized Cd36−/− female mice. (A) Naive Cd36−/− female mice (n = 5) were bred with WT male mice and delivered pups 3 times. The litter size was documented. One mother (#1) delivered a smaller littermate after the second and third pregnancy (litter size, 1.50 ± 0.71) compared with the other 4 mothers (#2-5) (litter size, 7.50 ± 1.93). Weak CD36-reactive antibodies were detected in mother #1 according to flow cytometry (see inset, the third delivery) but not in mothers #2 to #5 (data not shown). (B) Cd36−/− female mice (cohort #3) were immunized 3 times with WT platelets. Representative sera were then incubated with platelets from WT (white curves) or Cd36−/− mice (gray curves). Bound antibodies were detected with fluorescence-labeled anti-mouse IgG and analyzed by using flow cytometry. The percentages of positive cells (anti-CD36) are given. Sera from naive Cd36−/− were used as controls. (C) After immunization, Cd36−/− female mice (cohort #3; n = 16) were bred with WT male mice. Naive WT (cohort #1; n = 5) and naive Cd36−/− (cohort #2; n = 4) were conducted as controls. Dead pups were determined in utero or within 24 hours after delivery. The mortality of the pups in cohort #3 (31 of 77 [40.26%]) was compared with that of cohort #1 (χ2 = 17.46; ***P < .0001) and cohort #2 (χ2 = 11.52; ***P = .0007). (D) Severe bleeding in Cd36+/− pups delivered by immunized Cd36−/− mothers was found. (a) Healthy pup; (b) dead pup with bleeding (arrow); (c) dead pup with ICH (arrow); (d) dead pup with hydrops. (E) The litter size (including the dead pups) in naive WT, Cd36−/− mothers (cohorts #1 and #2), and immunized Cd36−/− mothers (cohort #3) is presented. The litter size in immunized Cd36−/− (cohort #3) was small but not significantly different compared with the naive WT (cohort #1) (4.81 ± 2.69 vs 7.40 ± 2.07; P = .064) and Cd36−/− (cohort #2) mothers (4.81 ± 2.69 vs 6.25 ± 1.50; P = .323) using a two-tailed unpaired Student t test.
The effect of maternal anti-CD36 antibodies on the fate of pups delivered by immunized Cd36−/− female mice. (A) Naive Cd36−/− female mice (n = 5) were bred with WT male mice and delivered pups 3 times. The litter size was documented. One mother (#1) delivered a smaller littermate after the second and third pregnancy (litter size, 1.50 ± 0.71) compared with the other 4 mothers (#2-5) (litter size, 7.50 ± 1.93). Weak CD36-reactive antibodies were detected in mother #1 according to flow cytometry (see inset, the third delivery) but not in mothers #2 to #5 (data not shown). (B) Cd36−/− female mice (cohort #3) were immunized 3 times with WT platelets. Representative sera were then incubated with platelets from WT (white curves) or Cd36−/− mice (gray curves). Bound antibodies were detected with fluorescence-labeled anti-mouse IgG and analyzed by using flow cytometry. The percentages of positive cells (anti-CD36) are given. Sera from naive Cd36−/− were used as controls. (C) After immunization, Cd36−/− female mice (cohort #3; n = 16) were bred with WT male mice. Naive WT (cohort #1; n = 5) and naive Cd36−/− (cohort #2; n = 4) were conducted as controls. Dead pups were determined in utero or within 24 hours after delivery. The mortality of the pups in cohort #3 (31 of 77 [40.26%]) was compared with that of cohort #1 (χ2 = 17.46; ***P < .0001) and cohort #2 (χ2 = 11.52; ***P = .0007). (D) Severe bleeding in Cd36+/− pups delivered by immunized Cd36−/− mothers was found. (a) Healthy pup; (b) dead pup with bleeding (arrow); (c) dead pup with ICH (arrow); (d) dead pup with hydrops. (E) The litter size (including the dead pups) in naive WT, Cd36−/− mothers (cohorts #1 and #2), and immunized Cd36−/− mothers (cohort #3) is presented. The litter size in immunized Cd36−/− (cohort #3) was small but not significantly different compared with the naive WT (cohort #1) (4.81 ± 2.69 vs 7.40 ± 2.07; P = .064) and Cd36−/− (cohort #2) mothers (4.81 ± 2.69 vs 6.25 ± 1.50; P = .323) using a two-tailed unpaired Student t test.
To establish a murine model of anti-CD36–mediated FNAIT at the first pregnancy, we immunized Cd36−/− female mice 3 times with 108 WT platelets before breeding with WT male mice. This study designed 8 cohorts (#1-8) (supplemental Table 1 [available on the Blood Web site]; Figure 1). Naive WT (#1) and Cd36−/− female mice (#2) were conducted as control cohorts. Flow cytometry analysis showed that Cd36−/− mice (#3) developed anti-CD36 antibodies after immunization (Figure 1B). Interestingly, these antibodies also reacted with human Cd36+/+ platelets (supplemental Figure 2). These results also pointed out that besides anti-CD36, no other platelet-reactive antibodies are involved in our animal model. In contrast, anti-CD36 was undetectable in naive Cd36−/− and naive Cd36+/+ mice (cohorts #1-2). Notably, 31 (40.26%) of 77 pups that were delivered from 16 immunized Cd36−/− mothers were found dead: 3 in utero and 28 pups after deliveries (bleeding, ICH, and hydrops) (Figure 1C-D). The mortality of pups in this cohort was significantly higher than in cohort #1 (χ2 = 17.46; P < .0001) and cohort #2 (χ2 = 11.52; P = .0007). In total, only 2.70% to 4.00% of dead pups were documented in the control cohorts (#1-2). Furthermore, litter size numbers from immunized Cd36−/− mothers with detectable anti-CD36 antibodies (cohort #3) showed a trend to be smaller than control cohorts; however, it was not statistically significant compared with naive WT (P = .064) and Cd36−/− (P = .323) mothers (Figure 1E). This smaller litter size was probably related to 2 miscarriages found in cohort #3, which were not found in the control cohorts.
Analysis of anti-CD36 antibodies in immunized Cd36−/− mothers and their corresponding pups showed that maternal anti-CD36 antibodies could not only be detected in fetal sera (circulating IgG) but also on fetal platelets (platelet-binding IgG) (Figure 2A). In these mothers, the platelet counts in the surviving pups were significantly lower than in pups from naive Cd36−/− mothers (377.12 ± 121.14 ×109/L vs 607.76 ± 87.80 ×109/L; P < .0001) (Figure 2B). A significant correlation (95% CI, 0.1161-0.8349; P = .0189) between anti-CD36 antibody titer and pups’ mortality rates was observed (Figure 2C).

Maternal anti-CD36 antibodies crossed the placenta and caused thrombocytopenia and fetus death. (A) Flow cytometry analysis: (a) circulating anti-CD36 IgG from an immunized Cd36−/− mother (white curve) was analyzed by using normal mice sera as control (gray curve). (b) Circulating anti-CD36 in the pups’ serum was measured by using pups’ serum from the naive mother as control (black curve). (c) Anti-CD36 IgG bound to pups’ platelets (platelet-binding IgG) was analyzed by using normal pups’ platelets from the naive mother as control (dark gray curve). (B) Platelet counts of pups from naive and immunized Cd36−/− mothers were counted by flow cytometry using counting beads as standard. The pups’ platelets (29 among 46 survival pups) from Cd36−/− immunized mothers (cohort #3; n = 7) is significantly decreased compared with pups’ platelets (13 among 24 survival pups) from naive mothers (cohort #2; n = 3). Data are expressed as mean ± standard deviation. Significance (***P < .0001) was analyzed by using a two-tailed unpaired Student t test. (C) Sera from immunized Cd36−/− mothers (n = 16) were analyzed by using flow cytometry. The reactivity of anti-CD36 antibodies in each maternal serum as relative fluorescence intensity (geometric mean) related to the percentage of mortality (number of dead pups/total pups) in the respective mothers is presented. Significance was analyzed by Pearson analysis (P = .0189; 95% confidence interval, 0.1161-0.8349). MFI, median fluorescence intensity.
Maternal anti-CD36 antibodies crossed the placenta and caused thrombocytopenia and fetus death. (A) Flow cytometry analysis: (a) circulating anti-CD36 IgG from an immunized Cd36−/− mother (white curve) was analyzed by using normal mice sera as control (gray curve). (b) Circulating anti-CD36 in the pups’ serum was measured by using pups’ serum from the naive mother as control (black curve). (c) Anti-CD36 IgG bound to pups’ platelets (platelet-binding IgG) was analyzed by using normal pups’ platelets from the naive mother as control (dark gray curve). (B) Platelet counts of pups from naive and immunized Cd36−/− mothers were counted by flow cytometry using counting beads as standard. The pups’ platelets (29 among 46 survival pups) from Cd36−/− immunized mothers (cohort #3; n = 7) is significantly decreased compared with pups’ platelets (13 among 24 survival pups) from naive mothers (cohort #2; n = 3). Data are expressed as mean ± standard deviation. Significance (***P < .0001) was analyzed by using a two-tailed unpaired Student t test. (C) Sera from immunized Cd36−/− mothers (n = 16) were analyzed by using flow cytometry. The reactivity of anti-CD36 antibodies in each maternal serum as relative fluorescence intensity (geometric mean) related to the percentage of mortality (number of dead pups/total pups) in the respective mothers is presented. Significance was analyzed by Pearson analysis (P = .0189; 95% confidence interval, 0.1161-0.8349). MFI, median fluorescence intensity.
These findings showed that maternal anti-CD36 antibodies could cross the placenta, bind to fetal platelets, and induce severe FNAIT with a high frequency of fetal death.
Early treatment with IVIG prevents FNAIT caused by maternal anti-CD36 antibodies
Subsequently, we queried whether IVIG could prevent the severe FNAIT found in our animal model. IVIG (1 g/kg) was intravenously administered to immunized Cd36−/− mothers 3 times on days 10, 15, and 20 after breeding. In the control experiment, no fetal death was detected in naive Cd36−/− mothers treated with IVIG (cohort #4) (Figure 3A). Surprisingly, this treatment did not prevent the severe clinical outcome of FNAIT. A high frequency of fetal death was still documented (40.00% vs 0.00%; P < .001) (cohort #5a). However, we found a significant reduction in fetal death (12.70% vs 40.00%; P < .01) when IVIG was administered earlier, on days 7, 12, and 17 (cohort #5b), although similar titers of maternal anti-CD36 antibodies before IVIG administration were observed in both cohorts (data not shown). In accordance, a reduced antibody titer was first observed on day 17 after the second antenatal administration of IVIG (P < .05) (Figure 3B).
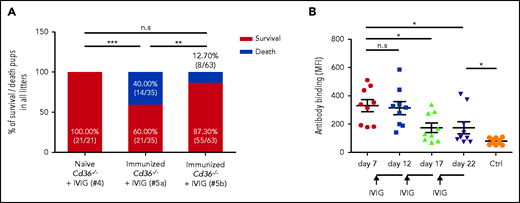
Early treatment with IVIG prevents FNAIT caused by maternal anti-CD36 antibodies. (A) Naive and immunized Cd36−/− female mice were bred with WT mice. During pregnancy, naive (cohort #4; n = 3) or immunized Cd36−/− mothers were treated with IVIG (1 g/kg) on days 10, 15, and 20 (cohort #5a; n = 7), or on days 7, 12, and 17 (cohort #5b; n = 9). High mortality of pups was observed in immunized mothers (cohort #5a) receiving IVIG on days 10, 15, and 20 compared with naive mothers (cohort #4) (P < .001). The number of dead pups was significantly decreased in immunized Cd36−/− mothers treated with IVIG 3 days earlier (cohort #5b), on days 7, 12, and 17 (P < .01). Significance was analyzed by using the χ2 test (**P < .01; ***P < .001). (B) Anti-CD36 antibodies in Cd36−/− immunized (cohort #5b; n = 9) were measured before (on day 7) and after (on days 12, 17, and 22) IVIG administrations by flow cytometry and are represented as median fluorescence intensity (MFI). Sera from naive mothers (Ctrl) were run as controls (n = 10). Data are expressed as mean ± standard deviation. Significance (*P < .05) was analyzed by using a two-tailed unpaired Student t test. n.s, not significant.
Early treatment with IVIG prevents FNAIT caused by maternal anti-CD36 antibodies. (A) Naive and immunized Cd36−/− female mice were bred with WT mice. During pregnancy, naive (cohort #4; n = 3) or immunized Cd36−/− mothers were treated with IVIG (1 g/kg) on days 10, 15, and 20 (cohort #5a; n = 7), or on days 7, 12, and 17 (cohort #5b; n = 9). High mortality of pups was observed in immunized mothers (cohort #5a) receiving IVIG on days 10, 15, and 20 compared with naive mothers (cohort #4) (P < .001). The number of dead pups was significantly decreased in immunized Cd36−/− mothers treated with IVIG 3 days earlier (cohort #5b), on days 7, 12, and 17 (P < .01). Significance was analyzed by using the χ2 test (**P < .01; ***P < .001). (B) Anti-CD36 antibodies in Cd36−/− immunized (cohort #5b; n = 9) were measured before (on day 7) and after (on days 12, 17, and 22) IVIG administrations by flow cytometry and are represented as median fluorescence intensity (MFI). Sera from naive mothers (Ctrl) were run as controls (n = 10). Data are expressed as mean ± standard deviation. Significance (*P < .05) was analyzed by using a two-tailed unpaired Student t test. n.s, not significant.
These results show that IVIG is suitable for the antenatal treatment of anti-CD36–mediated FNAIT, but particular care is required due to the delayed response of this therapy management.
deg-anti-CD36 antibodies prevent FNAIT caused by maternal anti-CD36 antibodies
Based on our previous studies, we sought to investigate whether deg-anti-CD36 antibodies could ease thrombocytopenia and prevent the severe effects of maternal anti-CD36 antibodies on the fetus during pregnancy.28 For this purpose, a pool of IgG purified from sera of immunized Cd36−/− mice (n = 10) and nonimmunized (n = 10; as control) mice were deglycosylated with PNGase F enzyme. Because undigested anti-CD36 IgG antibodies can worsen the pups’ fate, only pure deg-anti-CD36 was used for this in vivo study (supplemental Figure 3). When deg-anti-CD36 (5 mg/kg) was injected into immunized Cd36−/− mothers 3 times on days 10, 15, and 20 after breeding, the numbers of dead pups (cohort #7) were significantly lower than those of mothers treated with deg-mouse IgG (cohort #6) (5.26% vs 52.94%; P < .01) (Figure 4A).
![deg-anti-CD36 antibodies and deg-32-106 prevent FNAIT caused by maternal anti-CD36 antibodies. (A) Immunized Cd36−/− female mice were bred with WT mice. During pregnancy, Cd36−/− mothers were treated with deg-normal mouse IgG (cohort #6; n = 4), deg-anti-CD36 polyclonal antibodies (cohort #7; n = 3), or deg-32-106 (cohort #8; n = 6) in a dose of 5 mg/kg body weight on days 10, 15, and 20. The mortality of the pups treated with deg-anti-CD36 IgG (cohort #7; P < .01) or deg-32-106 (cohort #8; P < .0001) was significantly lower than that of mothers treated with deg-mouse IgG (cohort #6). Significance was analyzed by using the χ2 test (**P < .01; ***P < .001). (B) WT mice platelets were incubated with mAb 32-106 (black) and deg-32-106 (green) using isotype IgG as controls (gray). The binding of both antibodies was compared by using flow cytometry. (C) As indicated, 50 ng deg-normal mouse IgG (red), 50 ng deg-32-106 (blue), or 5 ng deg-32-106 (green) was incubated with WT mice platelets. After washings, platelets were incubated with fluorescence-labeled (fluorescein isothiocyanate [FITC]) maternal IgG containing anti-CD36 antibodies (13.8 µg IgG) and analyzed by flow cytometry. Fluorescence-labeled (FITC) mouse IgG (dotted line) was used as a negative control. Note the significant left shift of fluorescence intensity in the presence of deg-32-106 compared with normal mouse IgG. (D) Alexa Fluor–labeled deg-32-106 (1 mg/kg) was injected into WT female mice via the tail vein. After 10 minutes, FITC-conjugated maternal IgG containing anti-CD36 antibodies was administered. Subsequently, the binding of anti-CD36 and deg-32-106 antibodies was evaluated at 30 and 60 minutes. Note the decreasing frequency of FITC-labeled platelets (Q2 + Q3) when deg-32-106 was injected (54.8% vs 34.9%; 35.5% vs 14.9%). A single administration of labeled deg-32-106 or maternal IgG containing anti-CD36 was run as a control.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/18/10.1182_blood.2021011131/4/m_bloodbld2021011131f4.png?Expires=1770127970&Signature=qBXVW1lEjYPmIFqdQW3MljF9y~urwYo9xfUqxyJNpwiEz4M1ujaa6q4D3p5Z40nzayJilIyFbEGHSbdRAVsFfmWzMwTrpSLlfehrRn05v6Arstv4rcVJiKVG45wYJAqFUu7BRpVoryllg~X5P4lb3ld14vfa0dHdgevIepxmjBQfZzDj3m~Cs9uzlPyTu8fEM-3wEcF07BhANoajy-1l8~Remi2jw9tnVCCIwCUdvBL1~2rStIWpVqU2XxEa~FeShr5pGJIz6lYanTwK6-poOcka7i54bjWe6kS-di0rlRcZ58BadF5ZikBKgjDpqZoGqJ6vjEqx4RGJplZnYhD7qQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
deg-anti-CD36 antibodies and deg-32-106 prevent FNAIT caused by maternal anti-CD36 antibodies. (A) Immunized Cd36−/− female mice were bred with WT mice. During pregnancy, Cd36−/− mothers were treated with deg-normal mouse IgG (cohort #6; n = 4), deg-anti-CD36 polyclonal antibodies (cohort #7; n = 3), or deg-32-106 (cohort #8; n = 6) in a dose of 5 mg/kg body weight on days 10, 15, and 20. The mortality of the pups treated with deg-anti-CD36 IgG (cohort #7; P < .01) or deg-32-106 (cohort #8; P < .0001) was significantly lower than that of mothers treated with deg-mouse IgG (cohort #6). Significance was analyzed by using the χ2 test (**P < .01; ***P < .001). (B) WT mice platelets were incubated with mAb 32-106 (black) and deg-32-106 (green) using isotype IgG as controls (gray). The binding of both antibodies was compared by using flow cytometry. (C) As indicated, 50 ng deg-normal mouse IgG (red), 50 ng deg-32-106 (blue), or 5 ng deg-32-106 (green) was incubated with WT mice platelets. After washings, platelets were incubated with fluorescence-labeled (fluorescein isothiocyanate [FITC]) maternal IgG containing anti-CD36 antibodies (13.8 µg IgG) and analyzed by flow cytometry. Fluorescence-labeled (FITC) mouse IgG (dotted line) was used as a negative control. Note the significant left shift of fluorescence intensity in the presence of deg-32-106 compared with normal mouse IgG. (D) Alexa Fluor–labeled deg-32-106 (1 mg/kg) was injected into WT female mice via the tail vein. After 10 minutes, FITC-conjugated maternal IgG containing anti-CD36 antibodies was administered. Subsequently, the binding of anti-CD36 and deg-32-106 antibodies was evaluated at 30 and 60 minutes. Note the decreasing frequency of FITC-labeled platelets (Q2 + Q3) when deg-32-106 was injected (54.8% vs 34.9%; 35.5% vs 14.9%). A single administration of labeled deg-32-106 or maternal IgG containing anti-CD36 was run as a control.
deg-anti-CD36 antibodies and deg-32-106 prevent FNAIT caused by maternal anti-CD36 antibodies. (A) Immunized Cd36−/− female mice were bred with WT mice. During pregnancy, Cd36−/− mothers were treated with deg-normal mouse IgG (cohort #6; n = 4), deg-anti-CD36 polyclonal antibodies (cohort #7; n = 3), or deg-32-106 (cohort #8; n = 6) in a dose of 5 mg/kg body weight on days 10, 15, and 20. The mortality of the pups treated with deg-anti-CD36 IgG (cohort #7; P < .01) or deg-32-106 (cohort #8; P < .0001) was significantly lower than that of mothers treated with deg-mouse IgG (cohort #6). Significance was analyzed by using the χ2 test (**P < .01; ***P < .001). (B) WT mice platelets were incubated with mAb 32-106 (black) and deg-32-106 (green) using isotype IgG as controls (gray). The binding of both antibodies was compared by using flow cytometry. (C) As indicated, 50 ng deg-normal mouse IgG (red), 50 ng deg-32-106 (blue), or 5 ng deg-32-106 (green) was incubated with WT mice platelets. After washings, platelets were incubated with fluorescence-labeled (fluorescein isothiocyanate [FITC]) maternal IgG containing anti-CD36 antibodies (13.8 µg IgG) and analyzed by flow cytometry. Fluorescence-labeled (FITC) mouse IgG (dotted line) was used as a negative control. Note the significant left shift of fluorescence intensity in the presence of deg-32-106 compared with normal mouse IgG. (D) Alexa Fluor–labeled deg-32-106 (1 mg/kg) was injected into WT female mice via the tail vein. After 10 minutes, FITC-conjugated maternal IgG containing anti-CD36 antibodies was administered. Subsequently, the binding of anti-CD36 and deg-32-106 antibodies was evaluated at 30 and 60 minutes. Note the decreasing frequency of FITC-labeled platelets (Q2 + Q3) when deg-32-106 was injected (54.8% vs 34.9%; 35.5% vs 14.9%). A single administration of labeled deg-32-106 or maternal IgG containing anti-CD36 was run as a control.
Given the marked advantages of deg-anti-CD36 for the antenatal treatment of FNAIT, mouse mAbs against mouse CD36 were generated. One clone producing a high-affinity IgG2a antibody against mouse and human CD36, termed 32-106, was selected and deglycosylated. Flow cytometry analysis showed that both native and deg-32-106 IgG bound with similar affinity to WT platelets (Figure 4B). Furthermore, the competitive inhibition study in vitro (Figure 4C) showed that deg-32-106 could completely block the binding of maternal anti-CD36 antibodies into WT platelets. In vivo, administration of deg-32-106 significantly reduced the binding of maternal IgG containing anti-CD36 antibodies after 30 and 60 minutes (54.8% vs 34.9%; 35.5% vs 14.9%) (Figure 4D). However, the frequency of fluorescein isothiocyanate–labeled platelets (sensitized platelets) decreased after 60 minutes, most probably due to platelet clearance. Finally, we administered deg-32-106 (5 mg/kg) into immunized Cd36−/− mothers by the same therapy protocol (cohort #8). Only 1 dead pup (1 of 46 [2.17%]) was found in this cohort (P < .0001) (Figure 4A).
In addition, treatment with deg-32-106 led to increased pup platelet counts compared with the nontreated cohort (468.24 ± 85.20 ×109/L vs 377.12 ± 121.14 ×109/L; P < .05) (Figure 5A). Increased platelet counts (463.30 ± 30.88 ×109/L vs 377.12 ± 121.14 ×109/L; P < .05) were also observed with IVIG therapy; however, this was noted only when IVIG was administered early. These results showed that deg-32-106 could prevent the fatal severe effect of maternal anti-CD36 antibodies and improve the thrombocytopenic status of the fetus. More importantly, this treatment’s positive effect occurred more rapidly and was more effective compared with IVIG therapy. A similar phenomenon was observed with the litter size (Figure 5B).
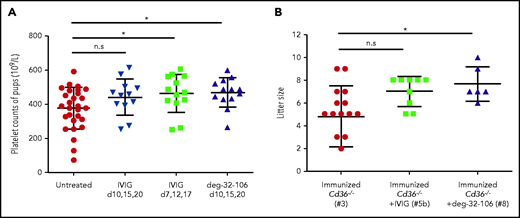
The effect of anti-CD36 on pups’ platelet counts and litter size from Cd36−/− immunized mothers treated with IVIG or deg-32-106 antibodies. (A) Platelets from survivor pups derived from untreated, IVIG-treated (1 g/kg), or deg-32-106–treated (5 mg/kg) mothers were collected and quantified by flow cytometry using counting beads as standard. The pups’ platelets from the untreated group were compared with those from 3 treated groups, including treatment with IVIG after breeding on days 10, 15, and 20; treatment with IVIG on days 7, 12, and 17; and treatment with deg-32-106 after breeding on days 10, 15, and 20. For each group, the platelets of 12 to 29 pups were counted. Data are expressed as mean ± standard deviation. Significance (*P < .05) was analyzed by using a two-tailed unpaired Student t test. (B) The litter size numbers derived from immunized Cd36−/− mothers treated with IVIG (cohort #5b; n = 9) or deg-32-106 (cohort #8; n = 6) were compared with those of untreated Cd36−/− mothers (cohort #3; n = 16). The data were analyzed by one-way analysis of variance, followed by the Bonferroni post hoc test (*P < .05). n.s, not significant.
The effect of anti-CD36 on pups’ platelet counts and litter size from Cd36−/− immunized mothers treated with IVIG or deg-32-106 antibodies. (A) Platelets from survivor pups derived from untreated, IVIG-treated (1 g/kg), or deg-32-106–treated (5 mg/kg) mothers were collected and quantified by flow cytometry using counting beads as standard. The pups’ platelets from the untreated group were compared with those from 3 treated groups, including treatment with IVIG after breeding on days 10, 15, and 20; treatment with IVIG on days 7, 12, and 17; and treatment with deg-32-106 after breeding on days 10, 15, and 20. For each group, the platelets of 12 to 29 pups were counted. Data are expressed as mean ± standard deviation. Significance (*P < .05) was analyzed by using a two-tailed unpaired Student t test. (B) The litter size numbers derived from immunized Cd36−/− mothers treated with IVIG (cohort #5b; n = 9) or deg-32-106 (cohort #8; n = 6) were compared with those of untreated Cd36−/− mothers (cohort #3; n = 16). The data were analyzed by one-way analysis of variance, followed by the Bonferroni post hoc test (*P < .05). n.s, not significant.
Placental dysfunction in FNAIT mediated by maternal anti-CD36 antibodies
Analysis of the placenta of immunized Cd36−/− mothers revealed a significant reduction in the placental labyrinth area compared with that of the naive cohort (55.75 ± 3.28% vs 62.88 ± 2.58%; P < .005) (Figure 6A-B). Furthermore, the quantification of vascular density in the placenta labyrinth by staining of endothelial cells with anti-CD31 antibodies showed lower fetal capillary numbers in the placenta labyrinth from immunized Cd36−/− mothers compared with naive cohort (9.86 ± 1.40 vs 13.98 ± 1.48; P < .0005) (Figure 6C-D).
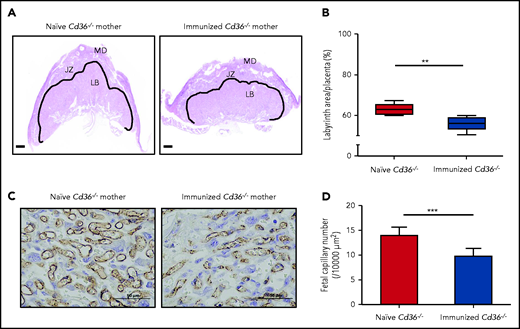
Anti-CD36 antibodies impaired vascularization in the immunized Cd36−/− mice placenta labyrinth zone. Additional 3 naive or Cd36−/− immunized female mice (n = 3) were crossed with WT male mice. The labyrinth zone was quantitatively analyzed at 16.5 days’ postcoitum. (A) The solid line shows the labyrinth area (scale bars, 500 µm). (B) The labyrinth ratio to the whole placenta is shown as a box-and-whisker plot. (C) Representative photomicrographs of the naive and immunized Cd36−/− placenta labyrinth areas stained with anti-CD31 antibodies. Scale bars, 50 µm. (D) Numbers of fetal capillaries in the labyrinth areas. Data are expressed as mean ± standard deviation . Significance (**P < .005; ***P < .0005) were analyzed by using a two-tailed unpaired Student t test. JZ, junctional zone; LB, labyrinthine layer; MD, maternal decidua.
Anti-CD36 antibodies impaired vascularization in the immunized Cd36−/− mice placenta labyrinth zone. Additional 3 naive or Cd36−/− immunized female mice (n = 3) were crossed with WT male mice. The labyrinth zone was quantitatively analyzed at 16.5 days’ postcoitum. (A) The solid line shows the labyrinth area (scale bars, 500 µm). (B) The labyrinth ratio to the whole placenta is shown as a box-and-whisker plot. (C) Representative photomicrographs of the naive and immunized Cd36−/− placenta labyrinth areas stained with anti-CD31 antibodies. Scale bars, 50 µm. (D) Numbers of fetal capillaries in the labyrinth areas. Data are expressed as mean ± standard deviation . Significance (**P < .005; ***P < .0005) were analyzed by using a two-tailed unpaired Student t test. JZ, junctional zone; LB, labyrinthine layer; MD, maternal decidua.
Subsequently, tube formation assay was performed by using the placenta microvascular endothelial cells (HPVEC) expressing human CD36 to study the influence of mouse sera containing anti-CD36 on the angiogenesis process. Our flow cytometry analysis showed that human CD36 could react with mAb 32-106 against mouse CD36 (supplemental Figure 4). As shown in Figure 7A to 7B, sera containing anti-CD36 antibodies significantly reduced the tube length compared with the control sera (P < .0005). A similar phenomenon was observed with 32-106. In contrast, deg-32-106 did not alter angiogenesis (Figure 7C-D), indicating the important role of FcγR. Indeed, blocking endothelial FcγRIIa with mAb IV.3 abolished the antiangiogenic effect of mAb 32-106 (supplemental Figure 5). Finally, preincubation of HPVEC with deg-32-106 restored the antiangiogenic effect caused by the anti-CD36 sera, both at 1:40 and 1:10 serum dilutions (P < .005). These results suggest that impaired angiogenesis of fetal placenta endothelial cells caused by maternal anti-CD36 antibodies could lead to the fetal death observed in our mouse model of FNAIT.
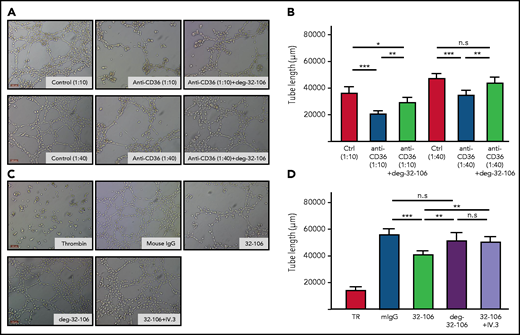
deg-32-106 prevented angiogenesis disturbance caused by the anti-CD36 antibodies. (A) HPVEC in Matrigel-coated wells were incubated with anti-CD36 sera (1:10, 1:40 dilution) in the absence or presence of deg-32-106. Diluted sera from naive mice were run as controls (control). (C) Thrombin (TR), 32-106, deg-32-106, or 32-106 together with mAb IV.3 against FcγRIIa was added to HPVEC as indicated. Mouse IgG was used as control. Scale bars, 100 µm. (B, D) Results from 3 experiments are presented. Data are expressed as mean ± standard deviation. Significance (*P < .05; **P < .005; and ***P < .0005) were analyzed by using a two-tailed unpaired Student t test.
deg-32-106 prevented angiogenesis disturbance caused by the anti-CD36 antibodies. (A) HPVEC in Matrigel-coated wells were incubated with anti-CD36 sera (1:10, 1:40 dilution) in the absence or presence of deg-32-106. Diluted sera from naive mice were run as controls (control). (C) Thrombin (TR), 32-106, deg-32-106, or 32-106 together with mAb IV.3 against FcγRIIa was added to HPVEC as indicated. Mouse IgG was used as control. Scale bars, 100 µm. (B, D) Results from 3 experiments are presented. Data are expressed as mean ± standard deviation. Significance (*P < .05; **P < .005; and ***P < .0005) were analyzed by using a two-tailed unpaired Student t test.
Discussion
This study established the first animal model of anti-CD36–mediated FNAIT and compared the capability of IVIG and deg-anti-CD36 antibodies to lessen FNAIT. After immunization with WT platelets, Cd36−/− female mice were bred with WT male mice. A high mortality rate of pups (40.26%) was detected. Furthermore, massive skin bleeding, hydrops fetalis, and ICH was identified. The surviving pups’ platelet counts (377.12 ± 121.14 ×109/L) were lower than those of the control cohorts. However, more severe thrombocytopenia (platelet counts, 132.2 ± 10.5 ×109/L) in pups caused by maternal anti-β3 antibodies was observed in a similar FNAIT model.29 The reason for this mild thrombocytopenia in our cohorts may be the low level of CD36 surface expression on mouse platelets (<25,000 copies) and the broad cellular distribution of the CD36 antigen (platelets, monocytes, macrophages, and endothelial cells).32 In contrast, high copy numbers of the αIIbβ3 integrin (110,000 to 130,000 copies/platelets) and the more restricted cellular distribution of mouse β3 integrin (platelets and endothelial cells) have been documented.33
In humans, variable clinical pictures of FNAIT caused by anti-CD36 antibodies have been observed, including widespread petechial hemorrhages, severe thrombocytopenia, ICH, and hydrops fetalis.10,15,16,20,34 All these clinical pictures were found in our FNAIT animal model, indicating that this model is suitable for studying the mechanism of anti-CD36–mediated FNAIT and proving certain therapy strategies.
Several treatment options have been conducted to prevent severe FNAIT caused by anti––HPA-1a antibodies, including serial fetal blood sampling, intrauterine platelet transfusions, and infusions of IVIG. A systematic review suggested that IVIG administration represents the first-line antenatal treatment, whereas fetal blood sampling and intrauterine platelet transfusion resulted in a high complication rate.1 Nevertheless, refractory states under IVIG treatment were still observed in mothers with ICH, although the success rate is 98.7%.35,36
Despite the increasing use of IVIG for treating FNAIT, the precise mechanism of IVIG action is still unclear.37 Several mechanisms have been proposed, including decreasing maternal antibody production by inducing immune tolerance38 and increasing pathogenic antibody clearance39 or decreasing antibody transport by saturated FcRn.40 In the β3−/− mice model of FNAIT, administration of IVIG could downregulate anti-β3 antibodies in both maternal and fetal circulations through FcRn-dependent and FcRn-independent pathways.3,29 However, such strategies may inhibit the transfer of immune-protective maternal IgG, which might cause an increased risk of infections by the local dominant micropathogens during pregnancy and in the first weeks following birth. Therefore, more specific and effective antenatal therapies are desirable.
Our previous studies have shown that deg-mAb SZ21 against HPA-1a could pass through the placenta, inhibit the binding of maternal anti–HPA-1a antibodies, and prevent the clearance of fetal platelets by macrophages.28 Furthermore, we found that some anti–HPA-1a antibodies explicitly bound to αvβ3 expressed by endothelial cells could induce endothelial dysfunction responsible for developing ICH in the fetus with severe FNAIT.41,42 More recently, we observed that deg-SZ21 could prevent not only thrombocytopenia but also inhibit endothelial dysfunction caused by anti-HPA-1a antibodies (S.S. manuscript in preparation). Based on this knowledge, an antenatal therapy for severe FNAIT based on epitope-specific competitive antibodies should be feasible.
Earlier studies have shown that most mAbs against CD36 and anti-CD36 sera recognized epitopes within amino acids 155-183, indicating the important role of this domain as an immune-dominant target for anti-CD36 antibodies.43,44 In the current study, we selected one mAb, 32-106, from our panel. Our in vitro and in vivo data showed that deg-mAb 32-106 could inhibit the binding of maternal anti-CD36 antibodies, indicating that both antibodies also react with the immune-dominant region. Accordingly, we found that the administration of deg-32-106 (5 mg/kg body weight) to the immunized Cd36−/− mothers 3 times (on days 10, 15, and 20) after breeding significantly increased not only fetal platelet counts (377.12 ± 121.14 ×109/L to 468.24 ± 85.20 ×109/L; P < .05) but also significantly reduced fetal death (40.26% to 2.17%; P < .005).
Surprisingly, similar antenatal treatment with IVIG administered in a dose of 1 g/kg body weight on days 10, 15, and 20 after breeding did not result in increased platelet counts and did not restore fetal death (40.00%). Based on risk stratification, weekly doses of IVIG (0.5 or 1 g/kg) are recommended to prevent bleeding complications in pregnancies complicated by FNAIT.37 At the start of treatment, the gestational age is mainly based on the estimated onset of ICH, ranging from 20 until 28 weeks of gestation. In women with a previous child with ICH, IVIG is commonly introduced earlier, at 12 weeks of gestation. This management indicates that early antennal treatment with IVIG prevents severe FNAIT. Following this view, IVIG administration 3 days earlier to the immunized Cd36−/− mothers (ie, days 7, 12, and 17) decreased fetal death (40.00% to 12.70%). However, fetal death frequency is still significantly higher than that of antenatal treatment with deg-32-106 (2.17%). The late response of IVIG may be attributed to the slow downregulation of maternal antibodies. In our animal model, downregulation of maternal anti-CD36 antibodies was observed only after the second administration of IVIG, which agrees with the current view mechanism that IVIG can prevent serious FNAIT by downregulation of maternal anti-CD36 titer due to clearance of IgG via FcRn.39 However, other mechanisms, such as direct inhibition of placenta transport by IVIG, should be considered.40 Nevertheless, the effect of IVIG seems to be delayed.
Furthermore, Leontyev et al45 reported that C57BL/6 mice are much less sensitive than BALB/c mice to IVIG-mediated attenuation of autoimmune thrombocytopenia (ITP), requiring ∼2.5-fold more IVIG (2.5 g/kg) than BALB/c mice. In the previous FNAIT model, mice on the BALB/c background were used to study the effect of IVIG on anti-β3 antibody-mediated FNAIT.3 Here, our results were based on the experiments with C57BL/6 mice, which may explain the low sensitivity of the IVIG treatment in our FNAIT model.
Disturbance of placenta vascular development and function could dramatically alter fetal growth development and thereby neonatal survival. In this process, placental vascularization and angiogenesis play critical roles.46,47 During the third trimester of pregnancy, placenta preferential transport of maternal plasma fatty acids is critical for fetal growth and development.48 CD36 is found on placental membranes, microvillus, and basal membrane.49 The central role of CD36 (also known as fatty acid translocase) as a high-affinity receptor for fatty acid uptake and lipid metabolism has been well documented.50,51 It is conceivable that the inhibition of fatty acid uptake via CD36 receptor by anti-CD36 antibodies could lead to inadequate placental angiogenesis. Indeed, the placenta analysis from immunized Cd36−/− mothers revealed a significant reduction of placental labyrinth area and decreased fetal capillary numbers compared with the naive cohort.
Previous studies reported on FcγRIIa (CD32) expression in the placenta microvascular endothelial cells.52 Accordingly, preincubation of HPVEC with mAb against FcγRIIa restored the antiangiogenic effect of anti-CD36 antibodies. Consequently, the decisive effect of anti-CD36 antibodies could be restored by the addition of deg-mAb 32-106. One study showed that antibodies bound to endothelial cells significantly increased polymorphonuclear leukocyte adhesion in an FcγRIIa-dependent manner in cooperation with CXCR1/2, a mechanism of tissue injury during the inflammatory response.53 The use of deg-32-106 may prevent antibody-mediated endothelial activation and recruitment of other blood cells such as monocytes and platelets. Taken together, our results showed that despite thrombocytopenia, maternal anti-CD36 antibodies contribute to fetal death by affecting placental angiogenesis.
In summary, we established a mouse model of FNAIT that reproduced the symptoms of human FNAIT induced by anti-CD36 antibodies. Although only mild thrombocytopenia was observed in the fetus, maternal anti-CD36 antibodies can cause severe bleeding, miscarriage, and fetal death. These severe clinical symptoms could be prevented by antenatal treatment with IVIG and deg-mAb 32-106 against CD36. Notably, treatment with deg-mAb against CD36 seems more beneficial than IVIG for various reasons, including the use of a lower dose (at least 200-fold less), the later start of treatment, and therapy success, despite other general IVIG disadvantages. Because our mouse hybridoma, 32-106, also recognizes human CD36 and can inhibit the binding of maternal anti-CD36 from FNAIT cases (supplemental Figure 6), humanized deg-mAb 32-106 should be feasible for immunotherapy in the near future.
Acknowledgments
The authors gratefully thank Jean-Pierre Allain (Department of Hematology, University of Cambridge) for his valuable suggestions and careful revision of the manuscript. They also thank LetPub (www.letpub.com) for its linguistic assistance and scientific consultation during the manuscript preparation.
This work was supported by grants from the National Natural Science Foundation of China (81601451 and 81970169), the Natural Science Foundation of Guangdong Province of China (2016A030313124 and 2020A1515011348), the Science and Technology and Innovative Commission of Guangzhou City (201707010021), and the Canadian Institute of Health Research Foundation Grant (389035) for H.N.
X.X. and D.C. are PhD candidates at the Justus Liebig University. This work is submitted in partial fulfillment of the doctoral requirements of the PhD.
Authorship
Contribution: X.X., X.Y., Y.F., and S.S. conceived and designed the research; X.X., D.C., Y.X., Y.C., Y.S., J.D., H.D., J.L., and J.W performed experiments; X.X., S.S., D.C., X.Y., and W.X. analyzed data; X.X., Y.F., and S.S. wrote the manuscript; and H.N. analyzed and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yongshui Fu, Guangzhou Blood Centre, 31 Lu Yuan Rd, Guangzhou, Guangdong, China 510095; e-mail: fuyongshui@sina.com; or Sentot Santoso, Institute for Clinical Immunology and Transfusion Medicine, Justus Liebig University Giessen, Langhansstr. 7, 35385 Giessen, Germany; e-mail: sentot.santoso@immunologie.med.uni-giessen.de.
All data-sharing requests may be submitted to the corresponding authors (Yongshui Fu [e-mail: fuyongshui@sina.com] or Sentot Santoso [e-mail: sentot.santoso@immunologie.med.uni-giessen.de]).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal