

In this issue of Blood, 1 report development of a preclinical model for anti-CD36–mediated fetal/neonatal alloimmune thrombocytopenia (FNAIT), providing novel insights on the pathogenic effect of maternal anti-CD36 antibodies. They also show that prenatal immunotherapy using both polyclonal and monoclonal murine anti-CD36 antibodies (mAbs) generated in the study was superior to human intravenous immunoglobulin (IVIg) treatment in preventing FNAIT.
CD36, also known as glycoprotein GPIV, is abundantly expressed on platelet membranes. It is also expressed by various other cell types, such as monocytes, erythroblasts, capillary endothelial cells, mammary cells, and epithelial cells and in membranes of the placenta. It is known to function as a receptor for various ligands, including collagen and thrombospondin-1, but, importantly, also for long-chain fatty acids, which mediate their uptake.
CD36 deficiency can manifest as either a complete deficiency across multiple cell types (type 1), or a partial deficiency affecting platelets only (type 2). Individuals with type 1 deficiency are at risk of isoimmunization upon CD36 exposure by transfusion or pregnancy, and such anti-CD36 antibodies can mediate platelet clearance, causing fetal/neonatal alloimmune thrombocytopenia (FNAIT), posttransfusion purpura, and platelet transfusion refractoriness. Whereas CD36 deficiency is rare in White populations, it is more frequent in African and Asian populations2,3 (type 1 reported in <2%), rendering isoimmunization to CD36 a significant cause of FNAIT in individuals from these ethnic groups.4
Currently, routine screening for platelet antigens to identify women at risk for alloimmunization is not preformed, and most neonates with FNAIT are identified at birth, prompting urgent laboratory investigations. Maternal CD36 deficiency can readily be identified by phenotyping platelets and monocytes using anti-CD36 antibody reagents in flow cytometry. Genotyping is not a straightforward diagnostic test because of the heterogeneous molecular basis of CD36 deficiency.4
In their work, Xu et al show that a commercially available CD36-knockout mouse strain develops platelet-reactive anti-CD36 antibodies after repeated challenges with wild-type platelets, and that breeding of such preimmunized females with wild-type males resulted in severe clinical disease in the heterozygous pups (see figure) Findings in this setting include increased mortality, prenatal bleeding, and thrombocytopenia in the surviving pups, in line with clinical outcomes in human pregnancies complicated by anti-CD36 antibodies.
Their comparative studies on placentas from immunized or nonimmunized Cd36−/− mice toward the end of pregnancy show that immunized mice with incompatible pregnancies had a smaller placental labyrinth area with reduced vascular density, presumably causing placental dysfunction. Proper vascularization of the murine placenta labyrinth is critical for fetal growth and development. Xu et al speculate that the observed reduction in vascular density is a result of placental angiogenesis that is hampered by pathogenic antibodies that bind to CD36 and thus impede fatty acid uptake by the placental tissue. Whether the reduced vascular density observed is a direct result of the antibody-blocking CD36, could have been investigated further by comparing placentas from mice carrying CD36-deficient pups, as the placental labyrinth phenotype depends on fetal genotype. With a suitable model at hand, Xu et al tested different immunotherapeutic options to prevent severe clinical FNAIT outcome. Intravenous immunoglobulin is often used (off-label) as an intervention in human alloimmunized pregnancies. It was also evaluated in the current study, with a moderate therapeutic effect on pup mortality when it was administered from early pregnancy.
For antigen-specific immunotherapy, antibody modifications are often necessary to yield the intended functionality. For human platelet antigen-1, which is the most commonly implicated antigen for FNAIT in White patients, different preclinical models show that antibody modifications (by deglycosylation or mutations) can turn an otherwise pathogenic antibody into a harmless effector silent variant suitable for prenatal immunotherapy.5-7 Xu et al identified a monoclonal anti-CD36 antibody (mAb 32-106), with high capacity for blocking CD36 binding in polyclonal sera. When used as prenatal immunotherapy, both deglycosylated, pooled polyclonal anti-CD36 sera and the deglycosylated monoclonal anti-CD36 antibody were shown to effectively increase pup survival and reduce FNAIT symptoms. Compared with IVIg, superior therapeutic effects were obtained with lower doses and with delayed intervention regimens.
What adds clinical appeal to this discovery is that mAb 32-106 also binds human CD36, providing an opportunity for translational research into the use of this antibody as prenatal immunotherapy for pregnancies complicated by maternal anti-CD36 antibodies. The most critical properties necessary to serve as a potential monoclonal therapeutic candidate for FNAIT are proper specificity and superior affinity, which block maternal pathogenic antibodies specific for the same antigen. For an isoantigen such as CD36, with tentatively multiple antigenic determinants, these may not be trivial issues. It is possible that a set of complementary mAbs may be needed. In light of this, the data presented on the ability of mAb 32-106 to efficiently compete for antigen with polyclonal human anti-CD36 FNAIT sera certainly is exciting. Although only 2 sera were tested, the result shows promise worthy of further study: can this lone monoclonal block the onslaught of a diverse polyclonal response? If this is the case, it may reveal new information regarding the immunogenic epitope(s) on CD36.
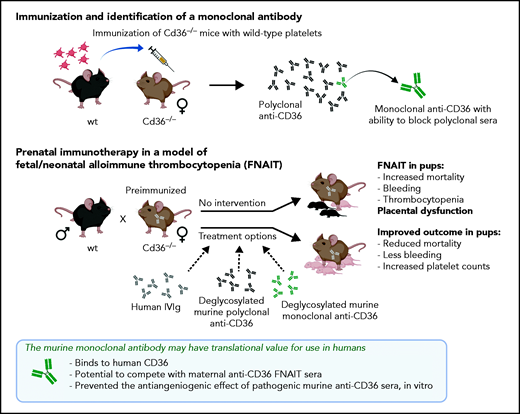
Cd36−/− mice generate anti-CD36 antibodies after repeated challenges with wild-type platelets. Breeding the preimmunized females with wild-type males generated placenta dysfunction and a severe FNAIT outcome in the pups. Pooled polyclonal sera and the mAb anti-CD36 identified by hybridoma selection were deglycosylated to remove effector function, to serve as a therapeutic antibody for inhibiting the binding of pathogenic maternal anti-CD36.
Cd36−/− mice generate anti-CD36 antibodies after repeated challenges with wild-type platelets. Breeding the preimmunized females with wild-type males generated placenta dysfunction and a severe FNAIT outcome in the pups. Pooled polyclonal sera and the mAb anti-CD36 identified by hybridoma selection were deglycosylated to remove effector function, to serve as a therapeutic antibody for inhibiting the binding of pathogenic maternal anti-CD36.
The road ahead, moving a murine monoclonal like mAb 32-106 into the clinic, is likely to involve a multistep process. Humanization will be needed to render it less immunogenic and to ensure appropriate binding to human FcRn, important for both transplacental transfer and longer half-life. In addition, abrogation of its effector function by targeted backbone mutations and potential affinity maturation before recombinant production should probably warrant consideration.
Different strategies to improve prenatal therapy for alloimmunized pregnancies and prevent FNAIT are in the pipeline. In addition to the off-label use of IVIg and therapeutic mAbs, FcRn-inhibitors have been suggested as a future treatment option for FNAIT, similar to ongoing clinical trials for red blood cell alloimmunized individuals.8 Although FcR inhibitors may offer a highly valuable 1-treatment-fits-all option for a wider range of implicated antigens, the advantage offered by targeting only the antigen of interest, while not affecting the half-life and transfer of maternal antibodies for passive immunity during pregnancy, is compelling.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal