

In this issue of Blood, report changes in histone posttranslational modifications (PTMs) and genome-wide RNA polymerase II occupancy during differentiation of primary human erythroid cells.1
Previous studies have shown that during erythropoiesis, RNA polymerase II (Pol II) transcription becomes more and more restricted to genes essential for the function of red blood cells.2 This is thought to be primarily the result of an overall decrease in chromatin accessibility and restricted recruitment of Pol II to erythroid genes by cell-type–specific transcription factors, including GATA1. Murphy et al demonstrate that although histone marks associated with active transcription (eg, H3 lysine 36 di- and tri-methylation [H3K36me2/3] and H3 lysine 79 di-methylation [H3K79me2]) decrease, heterochromatin-associated histone modifications (eg, H3 lysine 27 di- and tri-methylation [H3K27me2/3] and histone H3 lysine 9 di- and trimethylation [H3K9me2/3]) do not change during maturation of erythroid cells (see figure). Moreover, the authors demonstrate that elongation competent Pol II becomes limited during differentiation, which leads to an increased pausing index at genes not essential for the development of red blood cells. Erythroid-specific genes maintain efficient recruitment of elongation competent transcription complexes despite the marked reduction in Pol II. This study highlights an as yet underappreciated aspect of transcription regulation during erythropoiesis: the repression of transcription elongation at nonerythroid genes during a transient phase of erythroid maturation.
The authors used primary human CD34+ hematopoietic progenitor cells and induced differentiation along the erythroid lineage. At day 7, representing intermediate erythroblasts, and at day 10, representing mature erythroblasts, the cells were analyzed by mass spectrometry focusing on histone PTMs. The data show that histone PTMs associated with Pol II transcription elongation declined whereas PTMs typically associated with heterochromatin remained unchanged. ChIP-seq, ATAC-seq, and Cut and Tag experiments verified that decline in transcription activity was not associated with heterochromatin formation. Instead, the authors found that the decline in transcription of nonerythroid genes was the result of a defect in transcription elongation (at day 7) and eventually was the result of the lack of recruitment of Pol II (day 10). Remarkably, erythroid genes maintained high levels of transcribing Pol II. The finding that intermediate erythroblasts repress nonerythroid genes at the level of transcription elongation rather than at the level of chromatin accessibility or Pol II recruitment is unexpected and suggests a novel mechanism involved in erythroid cell specification. Intriguingly, the levels of activities that inhibit transcription elongation factor P-TEFb, HEXIM1, and the noncoding RNA 7SK, increase during erythroid maturation, with HEXIM1 exhibiting the highest levels in intermediate erythroblasts, at which stage the Pol II pausing index at nonerythroid genes is increased. The levels of P-TEFb decrease during erythropoiesis. Murphy et al also show that overexpression of HEXIM1 or HEXIM1 haploinsufficiency causes erythroid differentiation defects.
How do P-TEFb and HEXIM1 fit into the process of cell-specific gene regulation during erythropoiesis? After transcription initiation, Pol II pauses to allow capping of the 5' end of the RNA.3 This pause is mediated by negative elongation factors. P-TEFb phosphorylates these negative elongation factors as well as the Pol II C-terminal domain at serine 2, leading to the formation of an elongation competent transcription complex. How the reduced pool of transcriptionally active P-TEFb and elongation competent Pol II is preferentially recruited to erythroid genes becomes an important question, as emphasized by the Murphy et al study. P-TEFb consists of the CDK9 kinase and cyclin T1 or T2.4 HEXIM1 and 7SK RNA interact with the cyclin T1 subunit of P-TEFb. This renders P-TEFb unable to function as a transcription elongation factor. Several transcription factors have been shown to interact with cyclin T1 thereby dissociating HEXIM1 and allowing P-TEFb to activate transcription.4 For example, the HIV-encoded Tat protein efficiently competes with HEXIM1 for interactions with cyclin T and recruits active P-TEFb to paused Pol II in the HIV genome.5 With respect to hematopoietic cells, previous studies have shown that GATA1 and Ikaros transcription factors recruit P-TEFb to megakaryocytic and erythroid genes.6-8 This is consistent with the study by Murphy et al, which shows that elongation competent Pol II is preferentially recruited to GATA1-controlled genes, including genes involved in heme synthesis. These findings suggest that erythroid transcription factors including GATA1 either efficiently grab P-TEFb to erythroid genes in cells with declining Pol II activity, or that the HEXIM1/7SK RNA/P-TEFb complex is recruited to erythroid genes, and GATA1 displaces the negative components by interacting with cyclin T, thus exposing the CDK9 activity and allowing transcription to proceed.
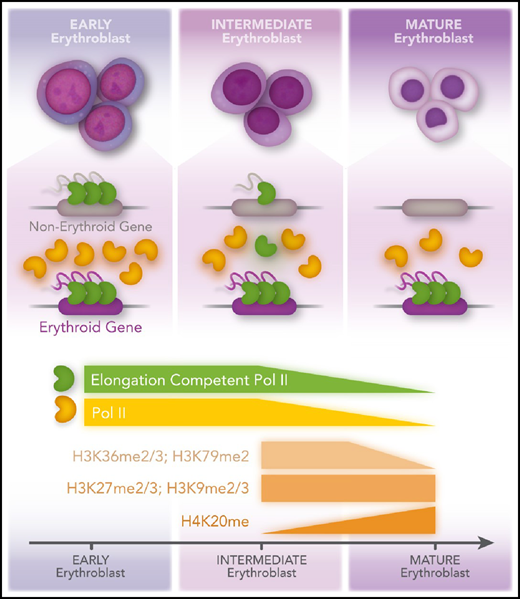
Murphy et al show that differentiation of erythroid maturation is accompanied by decreased histone posttranslational modifications associated with active transcription (H3K36me2/3; H3K79me2) but no change in repressive marks (H3K27me2/3; H3K9me2/3), except for H3K20me, which increases during maturation and is known to be associated with chromatin compaction in erythroid cells. In intermediate erythroblasts, there is an increased Poll II–pausing index at nonerythroid genes. In mature erythroblasts, Pol II levels decline, and recruitment is restricted to erythroid genes. Professional illustration by Patrick Lane, ScEYEnce Studios.
Murphy et al show that differentiation of erythroid maturation is accompanied by decreased histone posttranslational modifications associated with active transcription (H3K36me2/3; H3K79me2) but no change in repressive marks (H3K27me2/3; H3K9me2/3), except for H3K20me, which increases during maturation and is known to be associated with chromatin compaction in erythroid cells. In intermediate erythroblasts, there is an increased Poll II–pausing index at nonerythroid genes. In mature erythroblasts, Pol II levels decline, and recruitment is restricted to erythroid genes. Professional illustration by Patrick Lane, ScEYEnce Studios.
There are many anemias for which the molecular mechanism is unknown. As outlined by Murphy et al, GATA1 truncation mutations are associated with myeloproliferative disorders and Diamond-Blackfan–like anemias.9 The N-terminal truncation mutant GATA1s exhibits a similar chromatin occupancy profile compared with full-length GATA1 but fails to activate the erythroid-specific gene expression program.10 Therefore, GATA1s binds to chromatin but seems to be unable to recruit crucial activities that stimulate the recruitment and/or elongation activity of Pol II at erythroid-specific genes. It is thus tempting to speculate that the erythroid differentiation defect in cells expressing GATA1s or other GATA1 mutations is at least in part caused by the inability to recruit P-TEFb thereby converting Pol II into an elongation competent form at erythroid genes. However, the situation is likely more complex, because it was shown that GATA2 haploinsufficiency rescued the erythroid differentiation defect in GATA1s-mutant cells.10
In summary, the study by Murphy et al uncovers a hitherto unknown aspect of Pol II transcription regulation that will impact not only understanding of basic mechanisms driving erythroid cell maturation but may also uncover how specific GATA1 mutations negatively affect erythropoiesis and cause anemias.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal