

In this issue of Blood, ,1 report unexpected cell fate plasticity in erythroid progenitors (EPs) whereby removal or inhibition of the chromatin modifier LSD1 (also called KDM1a) triggers lineage conversion toward a myeloid fate, both in vitro and in vivo. This finding is important because it shows that progenitors that are typically thought of as being committed to the erythroid lineage have in fact retained the potential to differentiate toward other hematopoietic lineages. Furthermore, these findings provide strong support for the idea that active lineage restriction through epigenetic mechanisms, rather than irreversible loss of cell fate potential, underlies cell differentiation in hematopoiesis.
The search for strategies to reactivate fetal γ-globin genes expression in adult erythroid cells dates back to the discovery that even a small increase in the level of fetal hemoglobin is enough to significantly alleviate symptoms of β-globinopathies, including sickle cell disease and β-thalassemia.2 This early finding set the stage for decades-long research that led to the identification of multiple factors that inhibit fetal γ-globin genes expression, including transcriptional repressors (eg, BCL11A3) and chromatin-modifying enzymes. Although recent gene therapy trials targeting BCL11A have shown great promise for patients with sickle cell disease or β-thalassemia,4,5 the development of pharmacological agents to reactivate fetal hemoglobin remains a priority. A promising target for pharmacological induction of fetal hemoglobin is LSD1, an epigenetic enzyme that interacts with BCL11A and represses γ-globin genes transcription through the removal of active histone marks H3K4me1 and H3K4me2.6 Although previous studies showed that LSD1 inhibitors increase fetal hemoglobin levels in animal models,7 these chemicals also partly block erythroid differentiation, and the effects of prolonged loss of LSD1 activity in erythroid cells remain unknown.
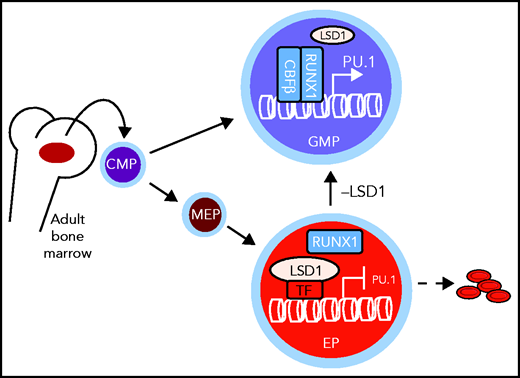
In committed adult EPs, differentiation toward the myeloid lineage is actively repressed by the chromatin-modifying enzyme LSD1. If LSD1 is depleted or inactivated pharmacologically, the cells convert to GMPs characterized by increased expression of the myeloid-specifying gene Pu.1. CMP, common myeloid progenitor; MEP, megakaryocyte-erythroid progenitor. See Figure 5F in the article by Yu et al that begins on page 1691.
In committed adult EPs, differentiation toward the myeloid lineage is actively repressed by the chromatin-modifying enzyme LSD1. If LSD1 is depleted or inactivated pharmacologically, the cells convert to GMPs characterized by increased expression of the myeloid-specifying gene Pu.1. CMP, common myeloid progenitor; MEP, megakaryocyte-erythroid progenitor. See Figure 5F in the article by Yu et al that begins on page 1691.
To address this question, Yu et al began by generating a new transgenic mouse model whereby a specific knockout can be induced exclusively in the erythroid lineage starting at the megakaryocyte-EP stage. By crossing this mouse with an LSD1 homozygous floxed mouse, they could then study the long-term consequences of LSD1 ablation in the adult erythroid lineage. First, they found that the mice were anemic, with significantly reduced numbers of early (blast-forming unit-erythroid [BFU-e]) and late (colony-forming unit-erythroid [CFU-e]) EPs. This result was expected, given the block of erythroid differentiation observed after LSD1 inhibition in cell culture. However, no increase in cell death was observed. In contrast, there was a significant increase in the number of granulocyte-macrophage progenitors (GMPs), suggesting that erythroid cells without LSD1 have converted to the myeloid lineage. This result was surprising, because “committed” EPs, as their name indicates, are thought to be incapable of a change in cell fate. To determine whether GMP-like cells are derived from LSD1-depleted EPs, Yu et al designed an elegant lineage-tracing experiment with a triple-mutant transgenic mouse, wherein only cells from the erythroid lineage were fluorescently marked with tandem tomato (TdT). Using this approach, they observed that GMP-like cells that arise in vivo upon LSD1 knockout are TdT+, which confirms that those cells originate from EPs. Importantly, they also showed that erythroid-derived GMP-like cells give rise to mature myeloid cells, which further supports the idea of an erythroid-to-myeloid switch upon LSD1 ablation. Further supporting this observation, they detected a reversal of the ratio between lineage-specifying transcription factors that control the erythroid (eg, GATA1) vs myeloid (eg, PU.1) cell fate transition and found increased binding of the myeloid transcription factor RUNX1 to the Pu.1 gene accompanied by enhanced H3K4me2 signal. Finally, they provided evidence that the erythroid-to-myeloid lineage conversion can be recapitulated in vitro using purified CFU-e cells exposed to an LSD1 inhibitor, which confirms that LSD1 acts through its enzymatic activity to restrict lineage potential in EPs (see figure).
Overall, the finding by Yu et al that cell fate decisions in committed EPs can be reversed by pharmacological inhibition of a chromatin-modifying enzyme has major implications for our understanding of the principles governing cell fate decisions and lineage commitment. Indeed, even though lineage commitment has been proposed to occur early in the hematopoietic stem/progenitor compartment (eg, Paul et al8 and reviewed in Watcham et al9), quantitative mass spectrometry experiments have indicated that transcription factors from nonerythroid lineages continue to be expressed in late EPs,10 which suggests that these cells still have the potential to differentiate toward other hematopoietic lineages. Consistent with this finding, Yu et al showed that EPs have not lost their potential to differentiate toward the myeloid lineage but rather that this potential is being actively suppressed by LSD1, at least partly through demethylation of H3K4me2. This finding implies that chromatin modifiers have an active role in restricting lineage potential and that, contrary to current beliefs, this process is reversible through epigenetic mechanisms. Another important finding is the decisive role played by myeloid transcription factors that continue to be expressed in EPs, in driving differentiation. Indeed, the deletion or inactivation of PU.1 or RUNX1 was sufficient to prevent myeloid conversion upon LSD1 inhibition. Overall, Yu et al provide important new evidence that lineage choice results from close cooperation between transcription factors that provide cells with the potential to differentiate toward multiple lineages and epigenetic modifications of chromatin that restrict this potential by regulating transcription factor activity and/or binding to lineage-determining genes.
Going forward, it is important to establish the full extent of chromatin changes (eg, H3K4me 1/2) and changes in transcription factors binding that occur upon LSD1 inhibition. Furthermore, the possibility that LSD1’s function in maintaining erythroid identity is also mediated through demethylation of nonhistone proteins remains to be addressed. Finally, the observation of cell fate plasticity upon LSD1 inhibition raises the possibility of increased susceptibility to malignant transformation by spurious activation of oncogenes, either through stochastic changes in gene expression or in response to environmental fluctuations. Regardless of the mechanism, malignant transformation should be carefully monitored when using pharmacological agents that can potentially alter cell fate decisions in the context of β-globinopathies or other diseases.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal