


Abstract
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common form of childhood cancer. Chemotherapy is associated with life-long health sequelae and fails in ∼20% of cases. Thus, prevention of leukemia would be preferable to treatment. Childhood leukemia frequently starts before birth, during fetal hematopoiesis. A first genetic hit (eg, the ETV6-RUNX1 gene fusion) leads to the expansion of preleukemic B-cell clones, which are detectable in healthy newborn cord blood (up to 5%). These preleukemic clones give rise to clinically overt leukemia in only ∼0.2% of carriers. Experimental evidence suggests that a major driver of conversion from the preleukemic to the leukemic state is exposure to immune challenges. Novel insights have shed light on immune host responses and how they shape the complex interplay between (1) inherited or acquired genetic predispositions, (2) exposure to infection, and (3) abnormal cytokine release from immunologically untrained cells. Here, we integrate the recently emerging concept of “trained immunity” into existing models of childhood BCP-ALL and suggest future avenues toward leukemia prevention.
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most frequent cancer in children and has a unique age peak at age 2-6 years. This age peak is distinct from other types of childhood leukemias, such as T-cell ALL (T-ALL) or acute myeloid leukemia (AML). Children with BCP-ALL have survival rates exceeding 90% after first- or second-line therapy, but treatment is intense and multimodal. Furthermore, survivors of BCP-ALL who have undergone therapy in childhood might suffer from later toxicity, morbidity, and mortality, a consequence that is a burden on the individual and the health care system in general.1,2 A deeper understanding of how preleukemic clones evolve to overt leukemia will be key in defining future preventive measures. Currently, there are 5 models of childhood leukemia evolution. Although the models differ in their specific mechanisms, they all point to infection-induced immune disturbances as being responsible for leukemia evolution.
In 1988, Kinlen proposed that leukemia is a rare consequence of exposure to a mild infectious agent in an isolated rural community suddenly faced with a rapid influx of newcomers eliciting novel immunological challenges. This so-called “population mixing theory” is based on analyses of leukemia incidence in rural and urban areas of Britain.3
At the same time, Greaves was the first who suggested that if an immature untrained immune system’s first infection exposure is delayed, the result is stronger, aberrantly damaging immune responses leading to the progression of preleukemic cells.4
Building on Greaves’ model, the “infective lymphoid recovery hypothesis” focuses on the leukemia-promoting effects of recurrent delayed infection-driven heat-shock responses and lymphoid involution early in life.5 Infections can lead to a release of pro-inflammatory (Th1) cytokines, which can in turn promote cell survival and a hypermutable state. In an attempt to restore cytokine homeostasis following infection, the release of Th2 cytokines and interleukin-7 (IL-7) then places a proliferative pressure on immature B cells, including preleukemic cells.
In contrast to the previous 3 models, Smith’s theory highlights the importance of in utero infections passed from mother to fetus.6 This model is supported by evidence from a large number of studies that were recently evaluated in a comprehensive systematic review and meta-analysis of maternal infection in pregnancy and childhood leukemia. The results showed a significantly increased BCP-ALL risk associated with in utero influenza, varicella, and rubella infections.7
Another theory, the adrenal hypothesis model by Schmiegelow et al,8 emphasizes the protective effect of early childhood infections that result in profound changes in the hypothalamus-pituitary-adrenal axis. Increased plasma cortisol levels resulting from infection-induced perturbations to the hypothalamus-pituitary-adrenal axis may directly eliminate preleukemic cells and suppress leukemia-promoting Th1-cytokine responses. This theory is supported by the fact that BCP-ALL patients are often extremely sensitive to glucocorticoid therapy.
In this review, we discuss how the absence of immune training early in life, as first proposed by Greaves, affects host responses to environmental challenges, and mechanisms by which this may promote BCP-ALL development. We further build on and refine these models by integrating the emerging novel concept of “trained immunity.” Trained immunity, in contrast to adaptive immune responses involving B and T cells, focuses on the responses and memory like properties of innate immune cells after infectious exposure and vaccination. This concept fits well into the time-restricted immune modulation occurring when children are particularly susceptible to BCP-ALL development. Finally, we summarize the current state of intervention research that ultimately aims to prevent the progression of the preleukemic clone into BCP-ALL.
Genetic predisposition in BCP-ALL
For childhood BCP-ALL to arise, a combination of genetic susceptibility and acquired somatic mutations is usually required.9 Genetic susceptibility observed in BCP-ALL is complex, ranging from very rare, but highly penetrant germline mutations in cancer predisposing genes to frequent, but low-penetrant somatic chromosomal aberrations and adverse combinations of germline single nucleotide polymorphisms associated with an elevated risk of developing childhood leukemia (Table 1).10 Most commonly, childhood BCP-ALL is characterized by recurrent somatic chromosomal aberrations, including aneuploidy and interchromosomal translocations11 originating in utero.9 These aberrations generate preleukemic cell clones, which frequently require secondary mutations to transform after a latency phase in early childhood (Figure 1).9 The most common translocation, t(12;21), encodes the oncogenic transcription factor ETV6-RUNX112 present in 5% of healthy newborns, ∼1 in 500 of which will develop the disease.13 Although systematic studies are lacking, it seems reasonable to speculate that healthy carriers clear preleukemic cells later in life. The age peak of BCP-ALL in children and the almost complete absence of the ETV6-RUNX1+ genetic subtype in adults with BCP-ALL, supports this view. There are few cases of ETV6-RUNX1+ BCP described in adults, and patients are usually young (median age, 24 years).14 In rare but informative cases of monozygotic twins (and thus complete HLA identity) with ETV6-RUNX1 predisposition, such silent, preleukemic cells can be detected years after birth without any evidence of disease.15
Genetic predisposition to BCP-ALL
| Rare, highly penetrant germline variations | ||||||
|---|---|---|---|---|---|---|
| Syndrome, gene(s) | Alteration | Consequence | Pathogenic variants | Presentation | Frequency | References |
| ETV6 | Missense, nonsense, frameshift, splice site, deletion | LOF, loss of transcriptional repression, probably dominant negative | Various, distributed throughout and clustered in DNA-binding Ets domain | Variable, thrombocytopenia, bleeding tendency, red cell macrocytosis, multilineage dysplasia, ∼1/3 have hematologic malignancies (ALL, MDS, AML). Solid tumors can occur in adulthood. | ∼1% of “sporadic” ALL | 107–114 |
| IKZF1 | Missense, nonsense, frameshift | LOF, altered subcellular localization, adhesion, and responsiveness to chemotherapy | Various, distributed throughout, mostly outside zinc finger regions | Immunodeficiency (CVID), autoimmunity, ALL | ∼1% of “sporadic” ALL | 115–119 |
| Li-Fraumeni syndrome, TP53 | Missense, nonsense, frameshift | LOF, decreased transcriptional activity | Various, distributed throughout and clustered in DNA binding | Osteosarcoma, breast cancer, soft-tissue sarcoma, brain tumors, adrenocortical carcinoma, ALL (mainly hypodiploid) | ∼0.5% of “sporadic” ALL | 109,120,121 |
| PAX5 | Missense | Hypomorphic variants, decreased repressive transcriptional activity | Arg38His Gly183Ser | ALL, no common abnormalities noted | Few affected families known | 122–125 |
| SH2B3 | Biallelic frameshift | Increased JAK-STAT signaling, accelerated proliferation of lymphoid cells | c.671insGGCCCCG p. Asp231Gly fs*38 | Mild developmental delay, growth retardation, autoimmunity, ALL | 2 siblings reported | 126 |
| TYK2 | Missense | GOF, promotes TYK2 autophosphorylation and activation of downstream STAT family members | p.Pro760Leu p.Gly761Val affecting the pseudokinase domain | ALL and second primary ALL | 2 unrelated patients reported | 127 |
| CMMRD syndrome, MLH1, MSH2, MSH6, PMS2 | Biallelic mutations | LOF | PMS2 c.1831dupA | Early-onset solid cancer and leukemia, café-au-lait spots, hypopigmented skin lesions, adenomatous polyps, pilomatricomas, or impaired immunoglobulin class switch recombination | ∼30% develop ALL or AML | 128,129 |
| Down syndrome (trisomy 21) | Trisomy, translocations | Aberrant gene dosage | Full trisomy of chromosome 21 or chromosome 21 translocations | Intellectual disability, cardiac abnormalities, facial dysmorphologies, transient abnormal myelopoiesis, predisposition to MDS, AML, ALL | ∼1% develop ALL or AML | 130 |
| Noonan syndrome, PTPN11, SOS1 | Missense, indels | GOF, dysregulate the RAS-MAPK pathway | PTPN11: SH2 domain, PTP domain interacting surfaces; SOS1: PH domain and distributed | Skin manifestations, growth retardation, facial dysmorphologies, cardiac abnormalities, neurofibroma, rhabdomyosarcoma, JMML, ALL, AML | ∼0.5% develop high hyperdiploid ALL | 131 |
| Rare, highly penetrant germline variations | ||||||
|---|---|---|---|---|---|---|
| Syndrome, gene(s) | Alteration | Consequence | Pathogenic variants | Presentation | Frequency | References |
| ETV6 | Missense, nonsense, frameshift, splice site, deletion | LOF, loss of transcriptional repression, probably dominant negative | Various, distributed throughout and clustered in DNA-binding Ets domain | Variable, thrombocytopenia, bleeding tendency, red cell macrocytosis, multilineage dysplasia, ∼1/3 have hematologic malignancies (ALL, MDS, AML). Solid tumors can occur in adulthood. | ∼1% of “sporadic” ALL | 107–114 |
| IKZF1 | Missense, nonsense, frameshift | LOF, altered subcellular localization, adhesion, and responsiveness to chemotherapy | Various, distributed throughout, mostly outside zinc finger regions | Immunodeficiency (CVID), autoimmunity, ALL | ∼1% of “sporadic” ALL | 115–119 |
| Li-Fraumeni syndrome, TP53 | Missense, nonsense, frameshift | LOF, decreased transcriptional activity | Various, distributed throughout and clustered in DNA binding | Osteosarcoma, breast cancer, soft-tissue sarcoma, brain tumors, adrenocortical carcinoma, ALL (mainly hypodiploid) | ∼0.5% of “sporadic” ALL | 109,120,121 |
| PAX5 | Missense | Hypomorphic variants, decreased repressive transcriptional activity | Arg38His Gly183Ser | ALL, no common abnormalities noted | Few affected families known | 122–125 |
| SH2B3 | Biallelic frameshift | Increased JAK-STAT signaling, accelerated proliferation of lymphoid cells | c.671insGGCCCCG p. Asp231Gly fs*38 | Mild developmental delay, growth retardation, autoimmunity, ALL | 2 siblings reported | 126 |
| TYK2 | Missense | GOF, promotes TYK2 autophosphorylation and activation of downstream STAT family members | p.Pro760Leu p.Gly761Val affecting the pseudokinase domain | ALL and second primary ALL | 2 unrelated patients reported | 127 |
| CMMRD syndrome, MLH1, MSH2, MSH6, PMS2 | Biallelic mutations | LOF | PMS2 c.1831dupA | Early-onset solid cancer and leukemia, café-au-lait spots, hypopigmented skin lesions, adenomatous polyps, pilomatricomas, or impaired immunoglobulin class switch recombination | ∼30% develop ALL or AML | 128,129 |
| Down syndrome (trisomy 21) | Trisomy, translocations | Aberrant gene dosage | Full trisomy of chromosome 21 or chromosome 21 translocations | Intellectual disability, cardiac abnormalities, facial dysmorphologies, transient abnormal myelopoiesis, predisposition to MDS, AML, ALL | ∼1% develop ALL or AML | 130 |
| Noonan syndrome, PTPN11, SOS1 | Missense, indels | GOF, dysregulate the RAS-MAPK pathway | PTPN11: SH2 domain, PTP domain interacting surfaces; SOS1: PH domain and distributed | Skin manifestations, growth retardation, facial dysmorphologies, cardiac abnormalities, neurofibroma, rhabdomyosarcoma, JMML, ALL, AML | ∼0.5% develop high hyperdiploid ALL | 131 |
| Frequent, low-penetrant germline variations in BCP-ALL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Location | Gene | dbSNP | Position | Risk allele | RAF | OR (95% CI) | Comments | References |
| 2q22.3 | Intergenic | rs17481869 | 2:145366886 | A | 0.03 | 1.74 (1.45-2.09) | ETV6-RUNX1 | 132,133 |
| 2p16.1 | Intergenic | rs2665658 | 2:60599667 | A | 0.34 | 4.0 (2.47-6.49) | TCF3-PBX1 | 134 |
| 3q28 | TP63, intronic, upstream | rs17505102 | 3:189683987 | G | 0.92 | 1.37 (1.25-1.48) | ETV6-RUNX1 | 135 |
| 5q31.1 | IRF1-AS1 intronic | rs886285 | 5:132429514 | T | 0.53 | 1.29 (1.18-1.41) | Hyperdiploidy | 133 |
| 6p21.31 | BAK1 intronic | rs210143 | 6:33579153 | C | 0.79 | 1.30 (1.19-1.43) | Hyperdiploidy | 133 |
| 7p12.2 | IKFZ1 intergenic | rs17133805 | 7: 50409816 | G | 0.21 | 1.65 (1.56-1-74) | — | 25,136–138 |
| 8q24.1 | CCDC26 intronic | rs4617118 | 8:129143897 | G | 0.21 | 1.28 (1.19-1.37) | — | 139 |
| 8q24.21 | CCDC26 intronic | rs75777619 | 8:129172930 | G | 0.12 | 1.26 (1.17-1.36) | — | 133 |
| 9p21.3 | CDKN2A p.Ala148Thr | rs3731249 | 9:21970917 | T | 0.01 | 2.23 (1.90-2.61) | — | 140–142 |
| 9q21.3 | CDKN2B-AS1 (intronic) | rs77728904 | 9:22057531 | C | 0.05 | 1.72 (1.50-1.97) | — | 143,144 |
| 10p14 | GATA3 intronic | rs3824662 | 10:8062245 | A | 0.20 | 1.29 (1.21-1.38) | Ph-like | 145,146 |
| 10p12.31-12.2 | PIP4K2A intronic | rs2296624 and others | 10:22568017 | C | 0.73 | 1.25 (1.18-1.32) | Hyperdiploidy, all ethnic groups, frequency: Hispanics > Europeans > Africans | 138,146,147 |
| 10q21.2 | ARID5B intronic, upstream variant | rs10821936 | 10:61963818 | C | 0.36 | 1.80 (1.71-1.89) | Hyperdiploidy, all ethnic groups, frequency: Hispanics > Europeans > Africans | 25,136–138 |
| 10q26.13 | LHPP | rs12779301 | 10:124604086 | C | 0.61 | 1.22 (1.15-1.29) | — | 144 |
| intronic | rs35837782 | 10:126293309 | A>C/G/T | |||||
| 11p11.2 | PTPRJ intronic | rs3942852 | 11:48093537 | T | 0.71 | 1.23 (1.11-1.32) | ETV6-RUNX1 | 135 |
| 12q23.1 | ELK3 intronic | rs4762284 | 12:96218984 | T | 0.43 | 1.15 (1.12-1.19) | — | 144 |
| 14q11.2 | CEBPE, SLC7A8, regulatory region | rs2239630 | 14:23120140 | A | 0.64 | 1.28 (1.22-1.35) | More frequent in Europeans | 25,148 |
| 17q12 | GSDMB intronic | rs2290400 | 17:39909987 | T | 0.58 | 1.18 (1.11-1.25) | — | 139 |
| 17q21.32 | IGF2BP1 Regulatory region | rs10853104 | 17:49014714 | T | 0.44 | 1.33 (1.21-1.47) | ETV6-RUNX1 | 133 |
| 21q22.2 | ERG intronic | rs9976326 | 21:38404563 | T | 0.22 | 1.33 (1.21-1.46) | High hyperdiploidy | 133 |
| 21q22.2 | ERG intronic | rs2836365 | 21:38396352 | G | 0.28 | 1.56 (1.33-1.83) | TCF3-PBX1, more frequent in Hispanics | 149 |
| Frequent, low-penetrant germline variations in BCP-ALL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Location | Gene | dbSNP | Position | Risk allele | RAF | OR (95% CI) | Comments | References |
| 2q22.3 | Intergenic | rs17481869 | 2:145366886 | A | 0.03 | 1.74 (1.45-2.09) | ETV6-RUNX1 | 132,133 |
| 2p16.1 | Intergenic | rs2665658 | 2:60599667 | A | 0.34 | 4.0 (2.47-6.49) | TCF3-PBX1 | 134 |
| 3q28 | TP63, intronic, upstream | rs17505102 | 3:189683987 | G | 0.92 | 1.37 (1.25-1.48) | ETV6-RUNX1 | 135 |
| 5q31.1 | IRF1-AS1 intronic | rs886285 | 5:132429514 | T | 0.53 | 1.29 (1.18-1.41) | Hyperdiploidy | 133 |
| 6p21.31 | BAK1 intronic | rs210143 | 6:33579153 | C | 0.79 | 1.30 (1.19-1.43) | Hyperdiploidy | 133 |
| 7p12.2 | IKFZ1 intergenic | rs17133805 | 7: 50409816 | G | 0.21 | 1.65 (1.56-1-74) | — | 25,136–138 |
| 8q24.1 | CCDC26 intronic | rs4617118 | 8:129143897 | G | 0.21 | 1.28 (1.19-1.37) | — | 139 |
| 8q24.21 | CCDC26 intronic | rs75777619 | 8:129172930 | G | 0.12 | 1.26 (1.17-1.36) | — | 133 |
| 9p21.3 | CDKN2A p.Ala148Thr | rs3731249 | 9:21970917 | T | 0.01 | 2.23 (1.90-2.61) | — | 140–142 |
| 9q21.3 | CDKN2B-AS1 (intronic) | rs77728904 | 9:22057531 | C | 0.05 | 1.72 (1.50-1.97) | — | 143,144 |
| 10p14 | GATA3 intronic | rs3824662 | 10:8062245 | A | 0.20 | 1.29 (1.21-1.38) | Ph-like | 145,146 |
| 10p12.31-12.2 | PIP4K2A intronic | rs2296624 and others | 10:22568017 | C | 0.73 | 1.25 (1.18-1.32) | Hyperdiploidy, all ethnic groups, frequency: Hispanics > Europeans > Africans | 138,146,147 |
| 10q21.2 | ARID5B intronic, upstream variant | rs10821936 | 10:61963818 | C | 0.36 | 1.80 (1.71-1.89) | Hyperdiploidy, all ethnic groups, frequency: Hispanics > Europeans > Africans | 25,136–138 |
| 10q26.13 | LHPP | rs12779301 | 10:124604086 | C | 0.61 | 1.22 (1.15-1.29) | — | 144 |
| intronic | rs35837782 | 10:126293309 | A>C/G/T | |||||
| 11p11.2 | PTPRJ intronic | rs3942852 | 11:48093537 | T | 0.71 | 1.23 (1.11-1.32) | ETV6-RUNX1 | 135 |
| 12q23.1 | ELK3 intronic | rs4762284 | 12:96218984 | T | 0.43 | 1.15 (1.12-1.19) | — | 144 |
| 14q11.2 | CEBPE, SLC7A8, regulatory region | rs2239630 | 14:23120140 | A | 0.64 | 1.28 (1.22-1.35) | More frequent in Europeans | 25,148 |
| 17q12 | GSDMB intronic | rs2290400 | 17:39909987 | T | 0.58 | 1.18 (1.11-1.25) | — | 139 |
| 17q21.32 | IGF2BP1 Regulatory region | rs10853104 | 17:49014714 | T | 0.44 | 1.33 (1.21-1.47) | ETV6-RUNX1 | 133 |
| 21q22.2 | ERG intronic | rs9976326 | 21:38404563 | T | 0.22 | 1.33 (1.21-1.46) | High hyperdiploidy | 133 |
| 21q22.2 | ERG intronic | rs2836365 | 21:38396352 | G | 0.28 | 1.56 (1.33-1.83) | TCF3-PBX1, more frequent in Hispanics | 149 |
| Frequent, prenatal, low-penetrant somatic variations in childhood BCP-ALL | ||||
|---|---|---|---|---|
| Variation | Alteration | Function | Frequency | References |
| ETV6-RUNX1 | Translocation | Chimeric transcription factor | ∼25% of BCP-ALL | 13,150 |
| High hyperdiploidy | Aneuploidy | Aberrant gene dosage | ∼25% of BCP-ALL | 151 |
| TCF3-PBX1 | Translocation | Chimeric transcription factor | ∼5-10% of BCP-ALL, frequency: Hispanics > Africans > Europeans | 152,153 |
| BCR-ABL1 | Translocation | Chimeric transcription factor | ∼3% | 154 |
| Frequent, prenatal, low-penetrant somatic variations in childhood BCP-ALL | ||||
|---|---|---|---|---|
| Variation | Alteration | Function | Frequency | References |
| ETV6-RUNX1 | Translocation | Chimeric transcription factor | ∼25% of BCP-ALL | 13,150 |
| High hyperdiploidy | Aneuploidy | Aberrant gene dosage | ∼25% of BCP-ALL | 151 |
| TCF3-PBX1 | Translocation | Chimeric transcription factor | ∼5-10% of BCP-ALL, frequency: Hispanics > Africans > Europeans | 152,153 |
| BCR-ABL1 | Translocation | Chimeric transcription factor | ∼3% | 154 |
CI, confidence interval; CMMRD, constitutional mismatch repair deficiency; CVID, common variable immunodeficiency; dbSNP, single nucleotide polymorphism database; GOF, gain of function; JMML, juvenile myelomonocytic leukemia; LOF, loss of function; MDS, myelodysplastic syndrome; OR, overall risk; RAF, risk allele frequency.
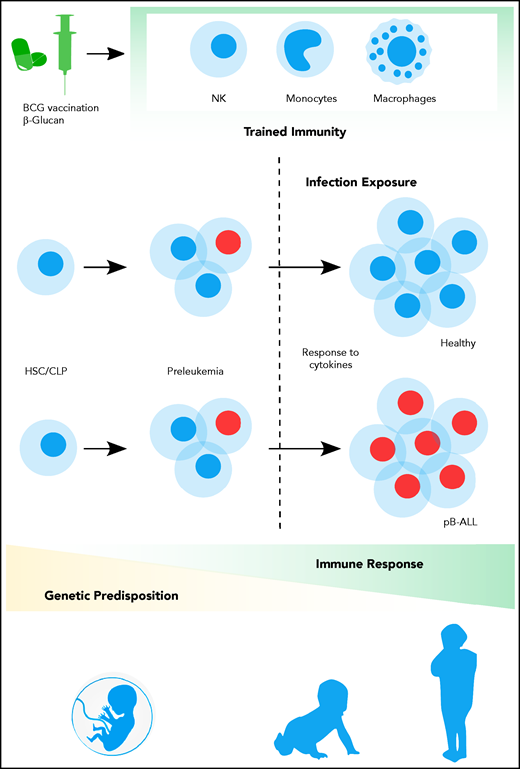
Contribution of trained immune responses to BCP-ALL development. Children genetically predisposed to BCP-ALL harbor clonally expanded preleukemic cells at birth. A hematopoietic stressor, such as infection, has the potential to trigger ALL at a later time point (2-6 years). The genetically determined immune responses, cytokine release, and basal cytokine levels, especially of interferons, may influence the outgrowth of the leukemic clone. However, the role of earlier-trained innate cells in the control of the preleukemic clone is largely unappreciated thus far. Epidemiological and experimental data suggest that innate immunity can be trained by BCG vaccination or β-glucan application, which substantially reduces the risk of developing BCP-ALL.
Contribution of trained immune responses to BCP-ALL development. Children genetically predisposed to BCP-ALL harbor clonally expanded preleukemic cells at birth. A hematopoietic stressor, such as infection, has the potential to trigger ALL at a later time point (2-6 years). The genetically determined immune responses, cytokine release, and basal cytokine levels, especially of interferons, may influence the outgrowth of the leukemic clone. However, the role of earlier-trained innate cells in the control of the preleukemic clone is largely unappreciated thus far. Epidemiological and experimental data suggest that innate immunity can be trained by BCG vaccination or β-glucan application, which substantially reduces the risk of developing BCP-ALL.
Infection exposure triggers BCP-ALL: evidence from epidemiology and preclinical mouse models
The age peak, first described by Ward,16 is strikingly unique to BCP-ALL and coincides with the period when children are commonly exposed to infections through interactions with their peers in day care, kindergarten, and primary school settings in developed societies.9 Ward proposed that infection acts as a trigger in childhood BCP-ALL.16 Epidemiological data linking leukemia occurrence to infection space-time clusters have supported this hypothesis (Table 2).17-19 Although childhood BCP-ALL generally does not cluster geographically,20 3 infection space-time clusters associated with increased BCP-ALL incidence may serve as examples: (1) Niles, USA (1957-1960)17; (2) Fallon, USA (1999-2004)18; and (3) Milan, Italy (2009-2010),19 the last of which was associated with an endemic AH1N1 swine flu outbreak. In Hong Kong in 2003, efforts to prevent the spread of communicable infections of severe acute respiratory syndrome (SARS) included a complete 2-month shutdown of public life, including schools and child day care facilities, and 6 months of additional strict measures. These actions resulted in a decrease in the number of common infections and coincided with a significant decrease in ALL.21
Selected epidemiological studies
| Space-time clustering of BCP-ALL | ||||
|---|---|---|---|---|
| Region | Associated agent | Cases | Time | References |
| Rural areas, UK | ND | NA | 1946-1965 | 3,155 |
| Niles, USA | Streptococcus | 8 | 1957-1960 | 17 |
| Fallon, USA | Adenovirus | 13 | 1999-2004 | 18 |
| Milan, Italy | Influenza A (H1N1) virus | 7 | 4 wk, 2009/2010 | 19 |
| UK | Influenza virus | NA | 1974-2000 | 156 |
| Switzerland | ND | NA | 1985-2014 | 157 |
| Space-time clustering of BCP-ALL | ||||
|---|---|---|---|---|
| Region | Associated agent | Cases | Time | References |
| Rural areas, UK | ND | NA | 1946-1965 | 3,155 |
| Niles, USA | Streptococcus | 8 | 1957-1960 | 17 |
| Fallon, USA | Adenovirus | 13 | 1999-2004 | 18 |
| Milan, Italy | Influenza A (H1N1) virus | 7 | 4 wk, 2009/2010 | 19 |
| UK | Influenza virus | NA | 1974-2000 | 156 |
| Switzerland | ND | NA | 1985-2014 | 157 |
| Proxies of exposure to infections associated with BCP-ALL | ||
|---|---|---|
| Proxy | Impact | References |
| Day care attendance | Increasing levels of social activity during the first year of life were associated with reduced risk. | 42 |
| Birth order | Being born later was associated with reduced risk. | 158 |
| Medication prescribed for infections | Medication prescribed for infections throughout childhood resulted in decreased risk. | 159 |
| Contact with livestock | Regular contact with livestock or pets was associated with lower risk. | 47 |
| Proxies of exposure to infections associated with BCP-ALL | ||
|---|---|---|
| Proxy | Impact | References |
| Day care attendance | Increasing levels of social activity during the first year of life were associated with reduced risk. | 42 |
| Birth order | Being born later was associated with reduced risk. | 158 |
| Medication prescribed for infections | Medication prescribed for infections throughout childhood resulted in decreased risk. | 159 |
| Contact with livestock | Regular contact with livestock or pets was associated with lower risk. | 47 |
| Immunological modifiers | ||
|---|---|---|
| Modifier | Impact | References |
| Mode of delivery | Increased risk was associated with cesarean section. | 36–38 |
| Breastfeeding | Breastfeeding for 6 mo or longer was associated with lower risk. | 40,88,89 |
| Birth weight | Higher birth weight was associated with increased risk. | 160,161 |
| Immunological modifiers | ||
|---|---|---|
| Modifier | Impact | References |
| Mode of delivery | Increased risk was associated with cesarean section. | 36–38 |
| Breastfeeding | Breastfeeding for 6 mo or longer was associated with lower risk. | 40,88,89 |
| Birth weight | Higher birth weight was associated with increased risk. | 160,161 |
mo, months; NA, not applicable; ND, not determined; wk, weeks.
It will be interesting to follow ALL incidence during the ongoing SARS-coronavirus-2 pandemic. Preliminary data were released by the Oslo University Hospital. They noted a reduction in ALL diagnoses in March 2020, when the Norwegian government implemented lockdown restrictions, closing schools, day care facilities, and after-school activities.22 The small sample size limits the conclusions that can be drawn, and cautionary notes were published to that effect.23
In summary, epidemiological studies suggest infection as a potential trigger of BCP-ALL in children.
These observations have been experimentally supported in vitro and in vivo (Table 3). Stimulation of ETV6-RUNX1+ transduced IL-7-dependent pre-B cells with bacterial lipopolysaccharide drove the expression of recombination activating gene 1/2 (RAG1/RAG2) and activation-induced cytidine deaminase (AID), as well as clonal evolution and outgrowth of BCP-ALL in a xenograft model.24 Another report found that genomic alterations caused by RAG1/RAG2 off-target activity, characterized by recombination signal sequence-like motifs near the breakpoints, dominated in patient- and clone-specific ETV6-RUNX1 fusions.25 Further reports showed that infection-induced RAG1/2- and AID-dependent genomic alterations24 and the composition of the hematopoietic niche, including the cytokine milieu26-28 and presence of innate immune cells,29 were critical to progression of preleukemia to BCP-ALL. ETV6-RUNX1+ cells demonstrated a competitive advantage in the presence of transforming growth factor-β compared with their wild-type counterparts.26 Furthermore, bone marrow stroma cells in the presence of tumor necrosis factor-α/IL-6 and IL-1β supported the outgrowth of ETV6-RUNX1+ preleukemic clones in a hematopoietic niche model of ETV6-RUNX1+ Ba/F3 cells.27
Preclinical murine BCP-ALL infection models
| Primary oncogenic lesion | Treatment | Outcome | Comment | References |
|---|---|---|---|---|
| Transgenic, retroviral LTR-driven ETV6-RUNX1 expression | No treatment | Decreased B-cell differentiation of early B-cell progenitors (Cd19− to pro-B) to pre-B cells | First model of ETV6-RUNX1 preleukemia | 162 |
| Transgenic, β-globin promoter–driven ETV6-RUNX1 expression, lymphoid lineage specificity via IGH chain enhancer | No treatment | Expansion of early B-cell progenitors (Cd34+Cd38−Cd19+) | First lymphoid lineage-specific model of ETV6-RUNX1 preleukemia | 26 |
| Heterozygous knockout, Pax5+/− | Exposure to infectious environment | BCP-ALL, ∼22% of mice | First in vivo model recapitulating human Pax5+/− BCP-ALL | 31 |
| Transgenic, retroviral LTR-driven ETV6-RUNX1 expression | NOD-SCID transplanted with pretreated Aicda+/+Rag1+/+ETV6-RUNX1 cells (IL-7 withdrawal, LPS treatment of AID activation) | 100% BCP-ALL in ex vivo LPS-treated Aicda+/+Rag1+/+ background | First murine model showing the impact of bacterial infection on ETV6-RUNX1+ leukemia development | 24 |
| Transgenic, Eμ-promoter-driven Ret expression | Treatment of IFNγ+/+ Eμ-ret mice with TLR ligands | Delay of BCP- ALL | First model of leukemia prevention through targeting IFN pathways | 29 |
| Transgenic, conditional E2A-promoter-driven E2A-PBX1 expression induced by Cd19-, Mb1-, or Mx1-driven Cre expression | No treatment | BCP-ALL: 7% Cd19-Cre line, 53% Mb1-Cre line, 59% Mx1-Cre line | First in vivo model recapitulating human E2A-PBX1 BCP-ALL | 163 |
| Transgenic, conditional E2A-promoter-driven E2A-PBX1 expression induced by Cd19-, Mb1-, or Mx1-driven Cre expression; Pax5+/− | No treatment | Heterozygous deletion of Pax5 substantially increased penetrance and shortened BCP-ALL latency | Confirmed a tumor-suppressive role for Pax5 in the TgE2A-PBX1 background | 163 |
| Transgenic, Sca1-promoter-driven ETV6-RUNX1 expression | Exposure to infectious environment | BCP-ALL, ∼10% of mice | First in vivo model recapitulating human ETV6-RUNX1+ BCP-ALL | 30 |
| Heterozygous knock-out, Pax5+/− Aid+/− | Exposure to infectious environment | BCP-ALL, ∼30% of mice | First model showing that AID does not affect latency or incidence of infection-mediated Pax5+/− BCP-ALL development | 32 |
| Hetero- and homozygous knock-out, Pax5+/− Aid−/− | Exposure to infectious environment | BCP-ALL, ∼30% of mice | ||
| Heterozygous knockout of Pax5+/− in heterozygous ν+ mice | Exposure to infectious environment | BCP-ALL, ∼15% of mice | First model showing that the infection-driven BCP-ALL development in Pax5+/− mice is not dependent on T cells | 81 |
| Heterozygous knockout Pax5+/− in homozygous ν/ν mice | Exposure to infectious environment | BCP-ALL, ∼15% of mice |
| Primary oncogenic lesion | Treatment | Outcome | Comment | References |
|---|---|---|---|---|
| Transgenic, retroviral LTR-driven ETV6-RUNX1 expression | No treatment | Decreased B-cell differentiation of early B-cell progenitors (Cd19− to pro-B) to pre-B cells | First model of ETV6-RUNX1 preleukemia | 162 |
| Transgenic, β-globin promoter–driven ETV6-RUNX1 expression, lymphoid lineage specificity via IGH chain enhancer | No treatment | Expansion of early B-cell progenitors (Cd34+Cd38−Cd19+) | First lymphoid lineage-specific model of ETV6-RUNX1 preleukemia | 26 |
| Heterozygous knockout, Pax5+/− | Exposure to infectious environment | BCP-ALL, ∼22% of mice | First in vivo model recapitulating human Pax5+/− BCP-ALL | 31 |
| Transgenic, retroviral LTR-driven ETV6-RUNX1 expression | NOD-SCID transplanted with pretreated Aicda+/+Rag1+/+ETV6-RUNX1 cells (IL-7 withdrawal, LPS treatment of AID activation) | 100% BCP-ALL in ex vivo LPS-treated Aicda+/+Rag1+/+ background | First murine model showing the impact of bacterial infection on ETV6-RUNX1+ leukemia development | 24 |
| Transgenic, Eμ-promoter-driven Ret expression | Treatment of IFNγ+/+ Eμ-ret mice with TLR ligands | Delay of BCP- ALL | First model of leukemia prevention through targeting IFN pathways | 29 |
| Transgenic, conditional E2A-promoter-driven E2A-PBX1 expression induced by Cd19-, Mb1-, or Mx1-driven Cre expression | No treatment | BCP-ALL: 7% Cd19-Cre line, 53% Mb1-Cre line, 59% Mx1-Cre line | First in vivo model recapitulating human E2A-PBX1 BCP-ALL | 163 |
| Transgenic, conditional E2A-promoter-driven E2A-PBX1 expression induced by Cd19-, Mb1-, or Mx1-driven Cre expression; Pax5+/− | No treatment | Heterozygous deletion of Pax5 substantially increased penetrance and shortened BCP-ALL latency | Confirmed a tumor-suppressive role for Pax5 in the TgE2A-PBX1 background | 163 |
| Transgenic, Sca1-promoter-driven ETV6-RUNX1 expression | Exposure to infectious environment | BCP-ALL, ∼10% of mice | First in vivo model recapitulating human ETV6-RUNX1+ BCP-ALL | 30 |
| Heterozygous knock-out, Pax5+/− Aid+/− | Exposure to infectious environment | BCP-ALL, ∼30% of mice | First model showing that AID does not affect latency or incidence of infection-mediated Pax5+/− BCP-ALL development | 32 |
| Hetero- and homozygous knock-out, Pax5+/− Aid−/− | Exposure to infectious environment | BCP-ALL, ∼30% of mice | ||
| Heterozygous knockout of Pax5+/− in heterozygous ν+ mice | Exposure to infectious environment | BCP-ALL, ∼15% of mice | First model showing that the infection-driven BCP-ALL development in Pax5+/− mice is not dependent on T cells | 81 |
| Heterozygous knockout Pax5+/− in homozygous ν/ν mice | Exposure to infectious environment | BCP-ALL, ∼15% of mice |
LPS, lipopolysaccharide.
These studies suggest additional overlaying mechanisms of leukemia evolution involving the interplay of preleukemic clones with innate immune and stromal cells in the bone marrow niche.27 In parallel, when transgenic mice with the ETV6-RUNX1 fusion30 or Pax5+/− heterozygosity31 were exposed to common infections, they developed BCP-ALL, although with incomplete penetrance. The high expression of AID observed in ETV6-RUNX1 preleukemic cells was recapitulated in Pax5+/− precursor cells, but did not affect BCP-ALL development.32 These murine models mimic specific aspects of BCP-ALL and enable the study of the interplay between genetic predisposition, host/environmental factors, and cooperating mutagenic events in BCP-ALL development. Notably, clonal evolution is not uniform in these murine models, but presents with distinct patterns of secondary somatic lesions dependent on the underlying genetic predisposition.
Evidence for training of immune cells in BCP-ALL
In contrast to lymphocyte-dependent immune responses, which lead to antigen-specific, long-term immunologic memories, the contributions of innate-like immune defenses have only recently gained attention. Challenging a long-standing dogma, it has become clear that cellular responses of innate immune cells are modified based on whether a previous encounter with infection or immune stressors had occurred. In the following sections, we review evidence for the contribution of proper and timely training of immune cells in BCP-ALL. Recent data collected from epidemiological, experimental, and clinical studies in mice and humans may pave the way for early intervention, hopefully even before clinically full-blown BCP-ALL develops.
Innate host immune responses influence penetrance of BCP-ALL
Epidemiological data demonstrating that infections have an inverse and protective effect early in life (<1 year of age) may initially appear to contradict what is known from space-time cluster data and preclinical mouse models. However, exposure to infectious agents and immune challenges by proxy in infancy reinforce the idea of an early infection-induced protective effect against ALL. Relevant factors include birth order,33-35 mode of delivery,36-39 breastfeeding,40 early day-care attendance,33,34,41-46 early common infection, and animal contact (reviewed in Ajrouche et al33). Further support for this hypothesis stems from a recent large-scale pooled and meta-analysis of 7847 leukemia cases (immunophenotype: 76% B-lineage, 10% T-ALL, rest unspecified/unknown) and 11 667 controls by the Childhood Leukemia International Consortium.47 The consortium demonstrated that regular contact with livestock, poultry, and pets in infancy (<1 year of age) reduced the risk of ALL development significantly.47 The reduced risk associated with contact with livestock was remarkably clear (odds ratio = 0.65; 95% confidence interval, 0.50-0.85).47 The influence of vaccines activating the innate and adaptive immune system, on the incidence of childhood ALL has also been explored. A meta-analysis of 12 studies48 observed that early vaccination (<3 months of age) with the Bacillus Calmette–Guérin (BCG) vaccine resulted in statistically robust protection from ALL.48 These associations are supported by numerous studies reporting on BCG vaccination of newborns and leukemia incidence in Austria,49 Chicago,50 and Quebec.51,52 In the latter 2 studies, the authors refer to mortality related to leukemia; thus, it remains debatable whether BCG vaccination modulated the course of leukemia or the mortality-associated infectious complications associated with the treatment. Before German reunification, BCG vaccination was compulsory in East but not in West Germany. This difference in vaccination protocol correlates with a lower rate of childhood leukemia in East Germany before reunification, which increased to West German levels 8 years after reunification.53,54
The protective role of early immune training in BCP-ALL development was explored in 2 transgenic murine BCP-ALL models: the Eμ-ret and the TCF3-PBX1 model.29,55 Toll-like receptors (TLRs) are pattern recognition receptors (PRRs) that detect potential harmful pathogens and activate downstream signaling pathways producing inflammatory cytokines (including type I interferon [IFN] and other mediators) that lead to the induction of innate immune responses. After ex vivo stimulation of TLR7, TLR8, or TLR9 leukemia-initiating precursor B cells derived from spleens of 4-week-old Eμ-ret mice showed reduced cell recovery, but increased cell expansion following TLR3 stimulation.29 Similar observations were made in the transgenic TCF3-PBX1 model.29,55 Treatment with IFN-α- or IFN-γ-neutralizing antibodies reversed these effects, implying that proliferation or regression of leukemia initiating cells is interferon-dependent.28,29 IFN-γ’s inhibitory activity on BCP-ALL was confirmed in IFN-γ−/− mice.28 Importantly, TLR9 stimulation induced long-term control of preleukemia and established leukemia in the same Eμ-ret model. Innate immune cells (namely natural killer [NK] cells and macrophages) were critical in mediating these effects.56 Collectively, these data provide a plausible mechanistic link between the reported association of early-life infections, immune modulation via PRRs, and protection from ALL. The long-lasting inhibitory effect was largely mediated by the innate rather than adaptive immune system, which points to the involvement of the innate immune cells’ memory.
Effects of trained immunity on host immune response and the hematopoietic stem cell compartment
Numerous studies laid the groundwork for establishing a new immunological principle, referred to as “innate immune memory” or “trained immunity,” to explain sustained memory-like properties of innate immune cells. In brief, macrophages, monocytes, and NK cells can undergo metabolic and epigenetic rewiring following exposure to infection, vaccination, or other immune stimuli, thereby modifying their expression profile and cell physiology. This plasticity provides innate immune cells with a memory, which subsequently modulates their response to a second, possibly heterologous stressor, such as infection exposure later in life.57 The induction of this long-lasting immunologic memory that is initiated at the level of bone marrow progenitors of the innate immune cells and is mediated by persistent epigenetic modifications in hematopoietic stem cells (HSCs) and myeloid progenitors depends on the transcription factor CCAAT/enhancer-binding protein β (C/EBPβ).58 Variations in individual responses to trained-immunity inducers have been explained by different DNA methylation patterns.59 Thus, when cells of the hematopoietic niche are trained with β-Glucan and BCG, they show accumulation of methylated and acetylated histone complexes, specifically H3K4me3 and H3K27ac. Kaufmann et al demonstrated an open accessible chromatin structure after BCG exposure in HSCs.60 Remarkably, these epigenetic marks were partially preserved when HSC differentiated along myeloid and lymphoid lineages. Further mechanistic studies may reveal how these marks are maintained during hematopoietic differentiation and remain stable through DNA replication and cell cycle. Thus, we advocate for studies addressing the link between epigenetic rewiring of innate immune cells and presumed changes in their methylation and acetylation status in pro- and anti-inflammatory cytokine loci, as well as its connection to clonal outgrowth of preleukemic clones.
To add complexity to the model, the individual composition of host microbiota, collectively referred to as the microbiome, also profoundly affects trained immunity responses. The microbiome functionally rewires bone marrow progenitors and adds to interindividual variation in cytokine responses.57,61,62 Transient infection and immune stimuli not only train innate immune cells but also functionally reshape bacterial species.
Stacy et al demonstrated that oral infection of wild-type mice with Klebsiella pneumoniae leads to long-term remodelling of intestinal microbiota and enhanced resistance to subsequent infection. They deciphered the functional metabolic relationships in these new defense processes.63 The infected host deploy more taurine, a bile-acid derived metabolite, as an essential nutrient and taurine-trained microbiota enhance colonization resistance.63
β-Glucan64 and live vaccines such as BCG65 are the best-studied inductors of trained immunity. β-Glucan, a component of cell walls in yeast, fungi, and seaweed, increases secretion of innate immune mediators such as IL-1β and granulocyte-macrophage colony-stimulating factor. Besides its ability to regulate infection,60,64 β-Glucan is approved as an immunoadjuvant therapeutic drug for cancer in Japan, Australia, South Korea, and Taiwan. Upon β-glucan-induced trained immunity, granulocyte-monocyte progenitors give rise to neutrophils with an anti-tumor phenotype and suppress tumor growth via production of reactive oxygen species. This phenomenon was accompanied by complete rewiring of granulopoiesis via transcriptomic and epigenetic changes.66 The anti-tumor activity crucially depended on IFN-I signaling because pharmacologic or genetic blockade of IFN-α/β receptor abolished the anti-tumor activity of trained immunity. The trained immunity effect was independent of adaptive immune cells, was long lasting and remained stable when trained neutrophils were systemically transferred into tumor-bearing mice.66 IFN-α is a key cytokine directing a multitude of context and time-dependent processes in the hematopoietic niche. Short-term, acute IFN-α stimulation of dormant HSCs leads to self-renewal and an activated state, whereas chronic IFN-α treatment blocks self-renewal and promotes progression to the progenitor state in vitro and in vivo.67 Thus, exposure to infection and the accompanying host IFN response directs proliferation and differentiation of HSCs/PCs. However, IFN-mediated effects on preleukemic or BCP-ALL cells likely elicit different responses. In a pluripotent hematopoietic stem/progenitor cell line (EML1) stably expressing ETV6-RUNX1, IFN-α/β production was suppressed following treatment with IL-7, thereby blocking B-cell differentiation at an early stage. The IFN-α/β pathway and IRF3 expression were suppressed in ETV6-RUNX1-expressing cells, but the differentiation block was relieved upon reexpression of IRF3, allowing cells to fully regain the capacity to differentiate into mature B cells.68
Cytokine profiles are altered at birth in children who later develop BCP-ALL
Two studies investigated cytokine levels at birth in children who subsequently developed BCP-ALL. Wiemels and colleagues measured 11 cytokines at birth in 116 childhood ALL cases and compared them with 116 healthy controls. Lower IL-10 levels were associated with an increased risk of developing ALL. IL-10 orchestrates the intensity and duration of immune responses and plays a complex context-specific role in tumor biology, although the cytokine itself has anti-tumoral properties and a pegylated version is being evaluated in clinical trials for the treatment of solid tumors.69,70 The second study showed that children who developed BCP-ALL had significantly lower neonatal concentrations of soluble IL-6 receptor (sIL-6Rα), IL-8, transforming growth factor β1, monocyte chemoattractant protein-1 (MCP-1/CCL2), and C-reactive protein, whereas concentrations of IL-6, IL-17, and IL-18 were significantly higher compared with controls.71 Overall, 8 of 9 detectable inflammatory markers in this study were abnormal in children who later developed BCP-ALL.
These studies suggest that children who develop BCP-ALL are born with an abnormal immune/cytokine response. Neither study assessed IFN levels, most likely because of technical limitations related to measuring these cytokines in dried neonatal blood spots. However, genome-wide studies have identified polymorphic IFNγ alleles associated with late onset of BCP-ALL in IFN-γ high producers and early onset in IFN-γ low producers, implying that in-born genetic polymorphisms determine the cytokine host response and affect BCP-ALL onset.72 This finding is further substantiated by the experimental observation that leukemia-initiating cell expansion was directly inhibited by IFN-γ but this phenomenon is restricted to the preleukemic phase only.28 The idea of inherited rare or common variants that affect the host response to infection exposure has been highlighted in recent years. The pattern of cytokine response related to specific pathogens in children is, to a large extent, inherited.73,74 This was shown in children both from healthy and diseased cohorts.75,76 Thus, in the future, cytogenomic studies deciphering host response to pathogens in specific leukemia subgroups will shed light on predisposing environmental conditions for the onset of leukemia. This approach will be an important tool for identifying children at risk early in life.
Overall, the complex interplay between three factors should be considered for the development of BCP-ALL: (1) inherited or acquired genetic risk factors (Table 1 and see Klco and Mullighan77); (2) exposure to infection; and (3) immunological immaturity with abnormal cytokine release of untrained cells (Figure 1). It seems plausible that the dysregulated cytokine profile is host-mediated, but not caused by the preleukemic cells themselves, since their frequency is very low.78,79
Toward intervention for prevention of BCP-ALL
Current knowledge points to several theory-guided, empirically supported avenues of BCP-ALL intervention and precautionary measures (Figure 2).
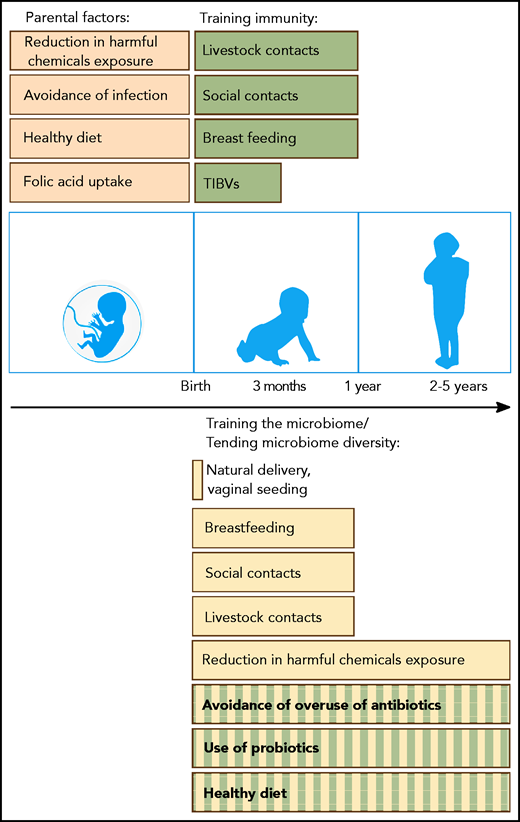
Possible preventive measures and proposed interventions that can help to reduce the risk of BCP-ALL development in genetically predisposed children. Before birth, maternal uptake of folic acid and a healthy diet (brown) have been associated with a reduced risk of BCP-ALL development. Maternal infection in pregnancy is associated with a significantly increased BCP-ALL risk related to viral transmission. After birth, trained immunity (green) and microbiome diversity (yellow) are important factors supported by epidemiological (filled bars) or experimental (striped bars) evidence. Immunity can be trained through vaccinations (TIBVs) before the age of 3 months, by breastfeeding and by social and livestock contacts (including pets) in the first year of life. Microbiome diversity is supported by a natural delivery and gradually builds up after birth. Again, breastfeeding and social and livestock contacts in the first year of life also have a beneficial impact on gut microbial diversity. Although only demonstrated in experimental models, the avoidance of overuse of antibiotics, the application of probiotics and a diet consisting of microbiome-supportive fibers are interventions that could also reduce the risk of leukemia development. Exposure of parents and children to various harmful chemicals can influence the microbiome along with carcinogenic effects.164-166 Further evidence needs to be generated through large population-based studies to identify preventive measures and to substantiate initial data on vaginal seeding and fecal transplants.
Possible preventive measures and proposed interventions that can help to reduce the risk of BCP-ALL development in genetically predisposed children. Before birth, maternal uptake of folic acid and a healthy diet (brown) have been associated with a reduced risk of BCP-ALL development. Maternal infection in pregnancy is associated with a significantly increased BCP-ALL risk related to viral transmission. After birth, trained immunity (green) and microbiome diversity (yellow) are important factors supported by epidemiological (filled bars) or experimental (striped bars) evidence. Immunity can be trained through vaccinations (TIBVs) before the age of 3 months, by breastfeeding and by social and livestock contacts (including pets) in the first year of life. Microbiome diversity is supported by a natural delivery and gradually builds up after birth. Again, breastfeeding and social and livestock contacts in the first year of life also have a beneficial impact on gut microbial diversity. Although only demonstrated in experimental models, the avoidance of overuse of antibiotics, the application of probiotics and a diet consisting of microbiome-supportive fibers are interventions that could also reduce the risk of leukemia development. Exposure of parents and children to various harmful chemicals can influence the microbiome along with carcinogenic effects.164-166 Further evidence needs to be generated through large population-based studies to identify preventive measures and to substantiate initial data on vaginal seeding and fecal transplants.
Ensuring microbiome diversity
The potentially fatal interplay between microbial signals and genetic leukemia predisposition has been demonstrated in Tet methylcytosine dioxygenase 2-deficient mice, whose preleukemic myeloproliferative state depends on microbially mediated inflammatory signals. Disruption of the intestinal barrier or stimulation of TLR2 agonists induced a preleukemic myeloproliferative state, whereas antibiotic treatment reverted this preleukemic condition.80 Diametrical effects of antibiotic treatment were investigated in a cohort of preleukemic Pax5+/− mice, in which we targeted the microbiome through treatment with an antibiotic cocktail over an 8-week period (Figure 3).81 Destruction and subsequent reconstitution of the gut microbiome triggered BCP-ALL in 50% of the mice, even when housed under specific pathogen-free conditions and lacking infectious stimuli.81 Untreated animals kept in the same specific pathogen-free animal housing facility remained healthy. These findings indicate a significant protective effect of the undisturbed, complex, and species-rich microbiome that is likely mediated through the release of microbial components or metabolites.
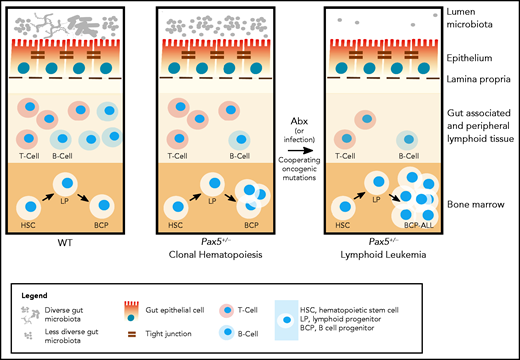
Antibiotic treatment in the development of lymphoblastic leukemia. Antibiotic treatment in early life induces leukemia in genetically predisposed Pax5+/− mice.81 (Left) In wild-type mice, depletion of the gut microbiome bacteria by antibiotic treatment at 8 weeks of age has only a transient effect on the immune system (including the gut-associated and peripheral lymphoid tissues) and mice do not develop pB-ALL. (Right) Pax5 heterozygosity directly affects B-cell maturation and leads to clonal hematopoiesis, while also indirectly reducing gut microbiota diversity. In response to bacterial depletion in the gut microbiome by antibiotic treatment at 8 weeks of age, the microbiome reconstitutes with further reduced diversity. Cooperating oncogenic mutations then lead to pB-ALL in ∼50% of these mice between 11 and 21 months of age. Leukemia development is preceded by a reduction of mature B and T cells in the gut and associated peripheral lymphoid tissues. However, it has not been tested in this model whether leukemia development can be inhibited through intervention. In addition to microbial dysbiosis, infectious stimuli can also cooperate with oncogenic mutations, leading to leukemia development in Pax5+/− mice.31
Antibiotic treatment in the development of lymphoblastic leukemia. Antibiotic treatment in early life induces leukemia in genetically predisposed Pax5+/− mice.81 (Left) In wild-type mice, depletion of the gut microbiome bacteria by antibiotic treatment at 8 weeks of age has only a transient effect on the immune system (including the gut-associated and peripheral lymphoid tissues) and mice do not develop pB-ALL. (Right) Pax5 heterozygosity directly affects B-cell maturation and leads to clonal hematopoiesis, while also indirectly reducing gut microbiota diversity. In response to bacterial depletion in the gut microbiome by antibiotic treatment at 8 weeks of age, the microbiome reconstitutes with further reduced diversity. Cooperating oncogenic mutations then lead to pB-ALL in ∼50% of these mice between 11 and 21 months of age. Leukemia development is preceded by a reduction of mature B and T cells in the gut and associated peripheral lymphoid tissues. However, it has not been tested in this model whether leukemia development can be inhibited through intervention. In addition to microbial dysbiosis, infectious stimuli can also cooperate with oncogenic mutations, leading to leukemia development in Pax5+/− mice.31
Notably, the gut microbiome of predisposed but still healthy Pax5+/− animals differed significantly from their wild-type counterparts long before leukemia onset.81
If confirmed in a human setting, microbiome testing may help to identify children at risk and provide a modifiable target for prevention. However, the data on microbiome diversity in healthy and genetically predisposed children are limited. Serial sampling from healthy children raises ethical and logistical questions, and, because of BCP-ALL’s low incidence, large-scale approaches would be required. So far, studies have only investigated the enteral microbiome in children at diagnosis, and during and after treatment of ALL. They found that the gut microbiome composition was age-dependent and predicted infection risk during chemotherapy,82 and that it persisted in a dysbiotic state years after chemotherapy.83,84 A difference in microbial composition was observed in survivors of ALL up to 11.9 years posttherapy.85 Microbial changes at diagnosis, however, are likely predominantly shaped by the disease and the accompanying perturbation of the immune system. We therefore advocate for future sampling approaches in healthy children on a population-wide scale. Another very heavily discussed field is vaginal seeding, or maternal-fecal microbiota transplantation in babies born by cesarean section. This procedure shifts microbiome composition toward a profile similar to that of babies born by vaginal delivery.86 Nevertheless, we believe that more research and a framework of well-controlled clinical trials are needed before the potentially beneficial effects of such a “bacterial baptism” procedure can be supported. Severe side effects, such as neonatal herpes-simplex infection, have been reported after vaginal seeding.87
Besides natural delivery and social contacts before 1 year, one of the most important factors in shaping the gut microbiome early in life is the mode of feeding: namely formula or breastfeeding. Comparing breastfed and formula-fed infants, both the intestinal microbiome and the lymphocyte population composition differed significantly, and the incidence of childhood leukemia was significantly lower in breastfed infants.40,88,89 A significantly greater amount of NK cells have been found in breastfed infants compared with formula-fed infants.90 Determining whether these correlative observations result in better NK cell-mediated surveillance of the preleukemic clone requires further experimental studies. However, the connection between several NK cell subsets, the expression of particular HLA-encoded ligands (C2) for inhibitory NK-cell receptors (KIR2DL) and increased susceptibility to BCP-ALL (but not T-ALL) has been demonstrated.91
Contact with livestock early in life not only fosters microbiome diversity, but also trains the immune system and significantly reduces the risk of BCP-ALL.47 In developed Western societies, the incidence of BCP-ALL is increased, as is that of asthma. These 2 disease states are epidemiologically linked and may represent 2 sides of the same coin.92 Both diseases are related to low exposure to immunological challenges in very early life. Of note, a thoughtfully designed birth cohort study recently demonstrated that the diversification of the gut microbiome of children growing up on a farm significantly contributed to asthma prevention.93 The protective effect was mainly mediated through specific microbial metabolites, such as fecal butyrate. Such studies can be used as a model toward prevention of BCP-ALL in children.
Training immunity
Immune responses in children are trained in early life through measures such as breastfeeding and social and livestock contact. A new concept of targeted intervention has recently emerged in form of trained-immunity-based vaccines (TIBV). Application of TIBVs seeks to increase host resistance against a broad spectrum of pathogens and to cross-protect against heterologous pathogens. Recently, TIBVs have been applied for prevention of autoimmune disease (including type 1 diabetes, multiple sclerosis), bladder cancer, and melanoma.94 TIBVs composed of PRR ligands are characterized by 2 distinguishing features that confer broad protection following administration. First, TIBVs aim to stimulate nonspecific effector responses of innate immune cells. Second, TIBVs stimulate the adaptive immune system through targeting activated dendritic cells, to increase antigen-specific and bystander responses. A TIBV example is the sublingual vaccine MV130 (composed of inactivated bacteria with a ratio of 90% gram-positive to 10% gram-negative strains), which is designed to prevent respiratory and urinary tract infections.95 It triggers dendritic cells to release the classical trained-immunity cytokines tumor necrosis factor α, IL-6, and IL-1β, leading to enhanced T-cell responses.96 In patients with common variable immunodeficiency, administration of MV130 resulted in a lower rate of respiratory infections, decreased antibiotic use, and fewer unscheduled doctor visits.97 MV130 also reduced the need for tonsillectomy in adults with recurrent tonsillitis.98 Well-studied TIBVs based on conventional vaccines are the BCG, Vaccinia, and influenza virus vaccines, which all can induce innate immune cell training.99 In a placebo-controlled clinical trial with attenuated yellow fever virus, healthy BCG-vaccinated volunteers showed a significant reduction in viremia and enhanced IL-1β production compared with the placebo group.100 BCG trained immunity effects are also beneficial in patients with non-muscle invasive bladder cancer as an immune-therapeutic approach in the urothelium, and have been used as a standard of care treatment of more than 40 years.101,102 In terms of efficacy, using a specific BCG strain is less important than the number of intravesical BCG installations. This principle is demonstrated by the NIMBUS randomized trial, in which patients who received a reduced number of BCG installations (n = 9 in the first year) showed far more cancer recurrences than patients treated with 15 installations.103 Furthermore, BCG vaccination of newborns reduced the risk of melanoma104 and leukemia, as reviewed previously. The protective link between BCG vaccination and childhood leukemia has been addressed by more than 12 studies since 1975.48 Although the protective beneficial effect of BCG as an immune modulator in early life is promising, little is known about potential detrimental effects. The protective mechanism of the trained immunity effect only lasts up to several months,105 although the capacity to enhance T-cell responses can be extended for up to 1 year.106 This is in sharp contrast to the sometimes life-long memory of adaptive immune cells gained through active infection. Thus, TIBVs are likely to have transitory rather than permanent effects, which narrows their therapeutic window and might require repetitive application. Given the sharp age peak of childhood BCP-ALL, as identified more than 100 years ago, we nevertheless envision that cross-protective effects of vaccines may have potential to be used for leukemia prevention. If well-controlled large-scale clinical trials prove the benefits of TIBVs or microbiome nurturing, such interventions may become a recommendation in pediatric care.
Summary
The reviewed data suggest the integration of trained immunity, with its key component of a temporary, unspecific immunological memory mediated by innate immune cells that lack the capacity to elicit antigen-specific responses, into the existing models of BCP-ALL development in children. The trained immunity concept, developed through epidemiological and genetic studies over the past several decades, adds a novel piece to the puzzle and provides a target for interventions. Immunity can be trained through the application of appropriate vaccinations early in life. Thus, adoption of the vaccination recommendation or immunity modulation via TIBVs, microbiome modulation, and avoidance of overuse of antibiotics will be promising avenues toward prevention of BCP-ALL in children.
Acknowledgments
The authors apologize for omitting some of the excellent published work related to the topic because of the limited number of references allowed. The authors thank Triantafyllos Chavakis, Martin Bornhäuser, Shai Izraeli, Aleksandra Pandyra, Stewart Boden, Daniel Hein, and Franziska Auer for useful suggestions and critical reading of the manuscript.
J.H. was supported by ERCStg 852222 “PreventALL,” ERAPerMed “GEPARD,” the German Cancer AID (Translational Oncology Program 70112951), and the German Jose Carreras Foundation (DJCLS 07/19). A.B. was supported by Löwenstern e.V., the Katharina-Hardt-Stiftung, and the German Carreras Foundation (DJCLS 07/19). A.B. and U.F. (FKZ: 3618S32275 and 3618S32274) were supported by the German Federal Office for Radiation Protection (BfS) and the Foundation Unoentrecienmil. U.F. was supported by the German Carreras Foundation (DJCLS 21R/2019).
Authorship
Contribution: J.H., U.F., and A.B. jointly contributed to conceptualization, screened and analyzed literature, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Arndt Borkhardt, Department for Pediatric Oncology, Hematology and Clinical Immunology, Medical Faculty, Heinrich-Heine University, Moorenstrasse, Düsseldorf 40225, Germany; e-mail: arndt.borkhardt@med.uni-duesseldorf.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal