

TO THE EDITOR:
Anti-CD19 chimeric antigen receptor (CAR) T-cell therapy induces high response rates and durable remissions in relapsed/refractory large B-cell lymphoma (LBCL).1 However, there is a need to elucidate relapse mechanisms to enable next-generation strategies. We evaluated CD19 antigen characteristics from pretreatment (N = 100) and postrelapse (N = 20) tumor biopsies, at protein and transcriptional levels, in patients with relapsed/refractory LBCL treated with axicabtagene ciloleucel (axi-cel; #NCT02348216) in the ZUMA-1 study and determined their association with clinical response.1,2 Of 20 relapsed biopsies, 18 had paired pretreatment and postrelapse biopsies tested using immunohistochemistry. Twenty-two pretreatment, 3 progression, and 6 paired biopsies also had sufficient tissue available for analysis by RNA sequencing. The study was conducted in accordance with the Good Clinical Practice guidelines of the International Conference on Harmonization. Additional information is provided in the supplemental methods, available on the Blood Web site.
Pretreatment biopsies showed variable CD19 and CD20 protein expression (H-score range, 0 to 300). Despite prior treatment with anti-CD20 antibody-containing chemotherapy in all patients, most biopsies pre–axi-cel showed CD20, alongside CD19 expression, and exhibited CD20 H-scores comparable or higher relative to CD19 (Figure 1A; supplemental Figure 1A-B). Reverse transcription polymerase chain reaction and western blot analysis confirmed variable CD19 messenger RNA and protein levels in CD19 CAR-T-naive LBCL tumors (supplemental Figure 1C-D; supplemental Table 1). CD19 H-scores pre–axi-cel were not significantly different across either best response (supplemental Figure 2A) or ongoing response groups (Figure 1B). Peak CAR T-cell levels post–axi-cel relative to tumor burden (TB), a correlate of clinical efficacy,3 were also comparable between pretreatment CD19 H-score groups across clinical response categories (Figure 1B).
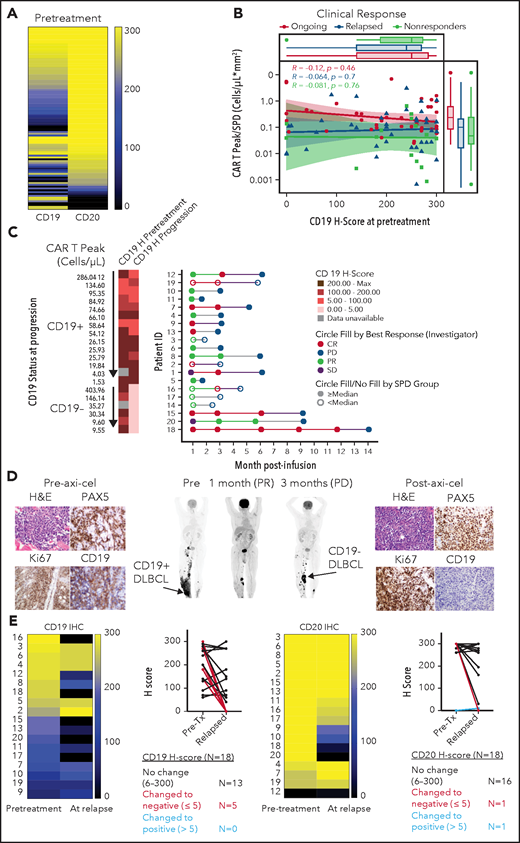
Association between CD19 levels at pretreatment or relapse and response. (A) Pretreatment CD19 H-score distribution in ZUMA-1 patients (N = 100) ordered by CD20 H-score: 90% CD19+, 93% CD20+, and 98% CD19+, and/or CD20+. CD19 and CD20 positivity was defined as H-score >5. (B) Association between pretreatment CD19 H-score with engraftment index (CAR T Peak/SPD) and clinical response. (C) Swimmer plot of relapse patients (N = 20) by CD19 status and peak CAR T-cell level. (D) Positron emission tomography scans and tumor biopsies from patient 21 at the indicated time points post–axi-cel. Brown staining shows positive signal from the respective immunohistochemistry marker with ×40 original magnification. Arrows show sites of tumor biopsy. (E) CD19 and CD20 H-scores in paired pretreatment-progression biopsies from relapsed patients (N = 18). CR, complete response; H&E, hematoxylin and eosin; PD, progressive disease; PR, partial response; SD, stable disease; SPD, sum of product of perpendicular diameters.
Association between CD19 levels at pretreatment or relapse and response. (A) Pretreatment CD19 H-score distribution in ZUMA-1 patients (N = 100) ordered by CD20 H-score: 90% CD19+, 93% CD20+, and 98% CD19+, and/or CD20+. CD19 and CD20 positivity was defined as H-score >5. (B) Association between pretreatment CD19 H-score with engraftment index (CAR T Peak/SPD) and clinical response. (C) Swimmer plot of relapse patients (N = 20) by CD19 status and peak CAR T-cell level. (D) Positron emission tomography scans and tumor biopsies from patient 21 at the indicated time points post–axi-cel. Brown staining shows positive signal from the respective immunohistochemistry marker with ×40 original magnification. Arrows show sites of tumor biopsy. (E) CD19 and CD20 H-scores in paired pretreatment-progression biopsies from relapsed patients (N = 18). CR, complete response; H&E, hematoxylin and eosin; PD, progressive disease; PR, partial response; SD, stable disease; SPD, sum of product of perpendicular diameters.
CD19− (vs CD19+) relapses were enriched in patients with lower pretreatment TB, most of which exhibit high CAR-T expansion (50% vs 21%) or in those experiencing the longest median time to relapse (9 vs 3 months; Figure 1C; supplemental Figure 2B-C). There was no difference in product attributes between CD19− and CD19+ relapses (supplemental Figure 2D). Importantly, in evaluable CD19+ relapse patients, peak expansion (and persistence) was lowest in patients with highest CD19 H-score at progression (Figure 1C; supplemental Figure 2E-F). These findings are consistent with higher selective pressure of axi-cel against CD19+ cells in a patient subset with increased CAR T-cell expansion and lowest TB and implicate 2 differential relapse mechanisms: (1) target-related through evasion with low/no CD19 tumor expression; and (2) suboptimal in vivo CAR T-cell expansion relative to TB potentially attributed to premature exhaustion, impaired trafficking, or immunosuppressive mechanisms in the tumor microenvironment (TME) contributing to CD19+ relapses.3 The latency of CD19− relapses in patients with higher pretreatment TB and low CAR T-cell expansion suggests a low frequency of CD19− cells, escaping under selective pressure after initial responses, potentially attributed to favorable TME.
Among 18 paired biopsies, 5 (28%) exhibited substantially lower CD19 protein levels at relapse (Figure 1D-E). CD19-low or negative relapses occurred after partial or complete responses and manifested at selected preexisting or new sites, suggesting heterogeneity in CD19 expression across different tumor sites within an individual (Figure 1D; supplemental Figures 3-4). Among 20 evaluable relapsed tumors (supplemental Table 2), most expressed other B-cell antigens (95% CD20, 90% CD22, and 95% CD79a), independent of CD19 expression level (supplemental Figures 5-6). However, greater heterogeneity was observed in CD22 protein expression vs CD20 and CD79a (supplemental Figure 5). Notably, several relapsed biopsies examined by immunofluorescence and confocal microscopy confirmed preservation of CD20 on the cell surface despite prior anti-CD20 antibody treatment (supplemental Figure 6), pointing to CD20 as an additional CAR target in LBCL. These data alongside prior studies4 suggest that multiantigen targeting could mitigate CD19 low or negative relapse. Finally, we observed pan absence of B-cell antigens at progression, on the background of myeloid gene enrichment in 1 relapse patient and 2 nonresponders (supplemental Figure 7). Although a myeloid switch has been reported in high-risk leukemia,5 we cannot rule out major myeloid cell infiltration into the TME as an alternate explanation to our findings.
Collectively, our data described above suggest that CD19− relapses likely emerge because of selective pressure after axi-cel. This contrasts with CD19+ relapses generally ascribable to lower product activity in vivo, manifested through reduced CAR T-cell expansion relative to pretreatment TB.3 Our data demonstrate that alternate B-cell antigens are frequently preserved independent of CD19 status at relapse.
Because the epitope for the anti-CD19 FMC63 antibody clone used in axi-cel generation is presumably a conformational epitope spanning exons 3 and 4,6,7 alternatively spliced exons (ASEs) and/or CD19 mutations may result in surface CAR-binding epitope loss and antigen evasion-mediated relapse without global impact on CD19 protein.8 Such genomic events are described in acute lymphoblastic leukemias relapsing after anti-CD19 CAR T-cell therapy8 but are not well characterized in LBCL. We observed that CD19 isoforms with partial or complete exon 2 and/or exons 5 to 6 deletions were present along with full-length CD19 isoforms in LBCL tumors not previously exposed to selective pressure with anti-CD19 CAR T cells (supplemental Figures 8-9; supplemental Tables 3-4) and, interestingly, even in normal B cells (supplemental Figure 10). Although exon deletions occur frequently, the data suggest that these splice variants coexist alongside full-length CD19 isoforms, without global loss of FMC63 binding epitope. Association between CD19 immunohistochemistry, detecting the cytoplasmic domain, and surface staining further support this notion (supplemental Figures 5-6).
Using the rMATS algorithm,9 we next analyzed RNA sequencing data from LBCL tumors in ZUMA-1 patients and found multiple ASEs in CD19 (Figure 2A). Interestingly, pretreatment samples showed enrichment of mutually exclusive exons in higher CD19 H-score biopsies (Figure 2B; supplemental Figure 11). Importantly, given the low number of events, all splice variants created by these events were likely subclonal, coexisting with the full-length CD19 isoform. Pretreatment CD19 transcript abundance was significantly associated with CD19 H-score but not with clinical response (Figure 2C).

Association between CD19 splice variants at pretreatment or relapse and response. (A) CD19 splicing events scheme. (B) Distribution of CD19 splice variants in pretreatment biopsies by CD19 H-score. (C) Association between pretreatment CD19 gene (transcript per million) and protein expression (left) and their respective relationship with ongoing response (middle and right). (D) Sashimi plots depicting characteristic ASEs in representative biopsies. (E) Prevalence of CD19 splice variants in paired pretreatment-relapse biopsies. (F) CD19 splice variants in relapse biopsies shown in panel E. Numbers denote impacted exons. (G) Response (left) and CD19 H-score (right) of patients shown in panel E. (H) Integrated Genomics Viewer snapshots showing CD19 mutations detected in paired pretreatment-relapse biopsies (representative patient 10). Blank, others; NR, nonresponder; O, ongoing response; Pt., patient; RE, relapsed.
Association between CD19 splice variants at pretreatment or relapse and response. (A) CD19 splicing events scheme. (B) Distribution of CD19 splice variants in pretreatment biopsies by CD19 H-score. (C) Association between pretreatment CD19 gene (transcript per million) and protein expression (left) and their respective relationship with ongoing response (middle and right). (D) Sashimi plots depicting characteristic ASEs in representative biopsies. (E) Prevalence of CD19 splice variants in paired pretreatment-relapse biopsies. (F) CD19 splice variants in relapse biopsies shown in panel E. Numbers denote impacted exons. (G) Response (left) and CD19 H-score (right) of patients shown in panel E. (H) Integrated Genomics Viewer snapshots showing CD19 mutations detected in paired pretreatment-relapse biopsies (representative patient 10). Blank, others; NR, nonresponder; O, ongoing response; Pt., patient; RE, relapsed.
In addition, analysis of 6 paired pretreatment-relapse and 3 unpaired progression samples revealed that ASEs and mutations are common, and CD19 transcript levels appeared lower in relapsed vs pretreatment tumors (Figure 2D-H; supplemental Figure 12A-B; supplemental Tables 5-8). These variants may be reflective of diverse tumor subclones, or alternatively, multiple isoforms may be present in any cell. Indeed, splicing events at relapse were not associated with clinical responses or CD19 H-score (Figure 2G; supplemental Figure 12C-D). ASEs and mutations that may alter the FMC63 epitope were more frequent at relapse vs pretreatment. However, coexpression of the full-length CD19 isoform in all cases and the low number of splicing (or mutation) events suggest that selection of unique epitope loss variants is unlikely to be a main mechanism of relapse. Schematic models of detected CD19 splice variants and mutations are provided (supplemental Figure 12G-H).9,10
In summary, CD19− relapse occurs in ∼30% of patients after axi-cel in LBCL likely because of indirect treatment-related selection of tumor cells with substantially low CD19 protein expression in the context of targeted antigen-positive tumor cell removal rather than ASEs or CD19 mutations. Our findings align with the ability of CD28-based CARs to control low antigen tumors.11-13 Alternate mechanisms underlying target-related evasion reported in the literature are disrupted CD19 cell membrane transport after endoplasmic reticulum,11,14 accidental insertion of CARs in tumor cells,15 or possible suboptimal bystander FAS-mediated killing of CD19− cells,16 further driving their selection. This mechanism of relapse complements other recently described mechanisms consisting of suboptimal product T-cell fitness and CAR expansion in vivo,3 or the role of immune TME-related factors that may influence outcomes.17 Together with evidence of preservation of alternate B-cell antigens at relapse in most tumors irrespective of CD19 levels, this work provides a rationale for multiantigen targeting in conjunction with optimizing T-cell product attributes to improve clinical outcomes.
Acknowledgments
The authors thank Francesca Milletti, Jenny J. Kim, Shruti Salunkhe, Stuart A. Sievers, Lianqing Zheng, Allen Xue, and Scott Turner for their contributions to this work. Medical writing support was provided by Ashley Skorusa and Skye Geherin of Nexus Global Group Science, funded by Kite, a Gilead Company.
This work was partly supported by the University of Texas MD Anderson Cancer Center B-cell Lymphoma Moonshot (S.S.N.) and National Institutes of Health, National Cancer Institute Cancer Center Support Grant P30 CA016672 to the University of Texas MD Anderson Cancer Center.
Authorship
Contribution: S.S.N., A.B., J.M.R., and V.P. developed the study design; V.P., J.C., L.W., S.P., G.H., Z.W., S.-Q.K., F.C., R.E.D., F.V., Z.B., C.A.J., F.L.L., P.M.R., S.J.R., L.J.L., I.W.F., and D.B.M. collected and analyzed the data; and all authors participated in data analysis, interpretation, manuscript writing, and approval of the final submitted version.
Conflict-of-interest disclosure: V.P. has employment with, stock or other ownership in, receives honoraria from, and receives travel support from Kite, a Gilead Company, and patents, royalties, or other intellectual property from Kite and Genentech. J.M.R. has employment with Kite, a Gilead Company. J.C. has employment with, stock or other ownership in, and receives travel support from Kite, a Gilead Company, and stock or other ownership in Five Prime Therapeutics. S.P. has employment with Kite, a Gilead Company; stock or other ownership in Gilead Sciences; and patents, royalties, or other intellectual property from UCLA. Z.W. has previous employment with Seattle Genetics; stock or other ownership with Seattle Genetics. Z.W. has prior employment with Seattle Genetics and current employment with Kite, a Gilead Company, and stock or other ownership in Seattle Genetics and Gilead Sciences. Z.B. has employment with Kite, a Gilead Company, and stock or other ownership in OmniacPharmConsult Ltd. C.A.J. has honoraria from Kite, a Gilead Company, Celgene, Novartis, Pfizer, Precision Biosciences, Nkarta, Lonza, and AbbVie; consultancy or advisory role for Kite, a Gilead Company, Novartis, Celgene, Bristol Myers Squibb, Precision Biosciences, Nkarta, and Lonza; speakers’ bureau participation for AXIS and Clinical Care Options; receives research funding from Pfizer; receives travel support from Kite, a Gilead Company, Novartis, Celgene, Bristol Myers Squibb, Precision Biosciences, Lonza, and Nakarta. F.L.L. has a scientific advisory role for Kite, a Gilead Company, Novartis, Amgen, Celgene/Bristol Myers Squibb, GammaDelta Therapeutics, Iovance, Bluebird Bio, Wugen, Calibr, and Allogene; is a consultant with grant options for Cellular Biomedicine Group, Inc; and receives research funding from Kite, a Gilead Company. P.M.R. has a consultancy or advisory role for Kite, a Gilead Company, and Curis, and receives research funding from Seattle Genetics. S.J.R. has stock or other ownership in Immunitas; receives research funding from Merck, Bristol Myers Squibb, Affimed, and Kite/Gilead; and receives travel support from Bristol Myers Squibb. I.W.F. has employment with Sarah Cannon Research Institute; stock or other ownership in Johnson & Johnson; serves as consultancy or advisory role for AbbVie, AstraZeneca, BeiGene, Curio Science, Gilead Sciences, Great Point Partners, Iksuda Therapeutics, Janssen, Juno Therapeutics, Kite, a Gilead Company, MorphoSys, Nurix Therapeutics, Pharmacyclics, Roche, Seattle Genetics, Takeda, Unum Therapeutics, Verastem, and Yingli Pharmaceuticals; receives research funding from AbbVie, Acerta Pharma, Agios, ArQule, AstraZeneca, BeiGene, Calithera Biosciences, Celgene, Constellation Pharmaceuticals, Curis, F. Hoffmann-la Roche, Forma Therapeutics, Forty Seven, Genentech, Gilead Sciences, IGM Biosciences, Incyte, Infinity Pharmaceuticals, Janssen, Juno Therapeutics, Karyopharm Therapeutics, Kite, a Gilead Company, Loxo, Merck, MorphoSys, Novartis, Pfizer, Pharmacyclics, Portola Pharmaceuticals, Roche, Seattle Genetics, Takeda, Teva, TG Therapeutics, Trillium Therapeutics, Triphase Research & Development Corp, Unum Therapeutics, and Verastem. D.B.M. has a consultancy or advisory role for Kite, a Gilead Company, Novartis, Juno-Celgene-Bristol Myers Squibb, Adaptive Biotech, Pharmacyclics, and Janssen; receives research funding from Kite, a Gilead Company, Novartis, Juno-Celgene-Bristol Myers Squibb, Adaptive Biotech, and Pharmacyclics; patents, royalties, or other intellectual property from Pharmacyclics; receives travel support from Kite, a Gilead Company, Novartis, Juno-Celgene-Bristol Myers Squibb, Adaptive Biotech, Pharmacyclics, and Janssen. A.B. has employment with Kite, a Gilead Company; stock or other ownership in, consultancy or advisory role for, and travel support from Gilead Sciences. S.S.N. has received personal fees from Kite, a Gilead Company, Merck, Bristol Myers Squibb, Novartis, Celgene, Pfizer, Allogene Therapeutics, Cell Medica/Kuur, Incyte, Precision Biosciences, Legend Biotech, Adicet Bio, Calibr, and Unum Therapeutics; research support from Kite, a Gilead Company, Bristol Myers Squibb, Merck, Poseida, Cellectis, Celgene, Karus Therapeutics, Unum Therapeutics, Allogene Therapeutics, Precision Biosciences, and Acerta; and patents, royalties, or other intellectual property from Takeda Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Sattva S. Neelapu, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: sneelapu@mdanderson.org.
The authors declare that the data supporting the findings of this study are available within the paper and its supplemental information files. Requests for raw data can be made through the corresponding author. Data can be found under accession number PRJNA727804.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal