

Key Points
N-glycan insertion within an extended epitope in the spacer domain prevents binding of pathogenic autoantibodies in iTTP.
Autoantibody-resistant NGLY3-ADAMTS13 (K608N) retains full proteolytic activity against von Willebrand factor.

Abstract
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is an autoimmune disorder caused by the development of autoantibodies targeting different domains of ADAMTS13. Profiling studies have shown that residues R568, F592, R660, Y661, and Y665 within exosite-3 of the spacer domain provide an immunodominant region of ADAMTS13 for pathogenic autoantibodies that develop in patients with iTTP. Modification of these 5 core residues with the goal of reducing autoantibody binding revealed a significant tradeoff between autoantibody resistance and proteolytic activity. Here, we employed structural bioinformatics to identify a larger epitope landscape on the ADAMTS13 spacer domain. Models of spacer-antibody complexes predicted that residues R568, L591, F592, K608, M609, R636, L637, R639, R660, Y661, Y665, and L668 contribute to an expanded epitope within the spacer domain. Based on bioinformatics-guided predictions, we designed a panel of N-glycan insertions in this expanded epitope to reduce the binding of spacer domain autoantibodies. One N-glycan variant (NGLY3-ADAMTS13, containing a K608N substitution) showed strongly reduced reactivity with TTP patient sera (28%) as compared with WT-ADAMTS13 (100%). Insertion of an N-glycan at amino acid position 608 did not interfere with processing of von Willebrand factor, positioning the resulting NGLY3-ADAMTS13 variant as a potential novel therapeutic option for treatment of iTTP.
Introduction
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is a rare life-threatening autoimmune disease.1-3 In iTTP, the von Willebrand factor (VWF) cleaving protease ADAMTS13 is targeted by autoantibodies,4 which leads to loss of its proteolytic activity5 and/or enhanced clearance6 of ADAMTS13. A major epitope (R568/F592/R660/Y661/Y665)4,7-9 for autoantibodies is located in the spacer domain exosite-3, which also provides an interaction hub for VWF.9,10 Early studies revealed that modification of exosite-3 resulted in impaired binding of autoantibodies.4,7 Conservative amino acid substitutions within exosite-3 yielded a so-called gain of function (GoF; R568K/F592Y/R660K/Y661F/Y665F) ADAMTS13 variant that conferred resistance to autoantibodies.8 In contrast to these findings, quantitative assessment of autoantibody binding using an extensive panel of conservative, semiconservative, and nonconservative exosite-3 variants indicated that conservative substitutions that included the GoF variant did not alleviate the binding of pathogenic autoantibodies.10 Semiconservative and nonconservative substitutions did result in a significant loss of autoantibody binding but also reduced proteolytic activity.10 In the current study, we employed structural bioinformatics to explore a larger epitope landscape on the ADAMTS13 spacer domain. Newly generated autoantibody-spacer domain complex models were used to design novel variants capable of resisting patient autoantibody binding. We show that artificially inserted N-glycans prohibit binding of pathogenic autoantibodies while retaining normal levels of proteolytic activity.
Study design
3D structural models of Fv (variable fragments) from patient-derived monoclonal anti-ADAMTS13 autoantibodies II-1 and I-95,11 were built (Figure 1A).12,13 The possible complex formations between ADAMTS13 cysteine-rich/spacer domain crystal structure (Protein Data Bank ID: 3GHM)14 and Fv models were generated by protein-protein docking (Figure 1B).15 Each docking pose for the wild-type (WT) antigen-autoantibody complex was also exploited to generate GoF ADAMTS13 mutations (R568K/F592Y/R660K/Y661F/Y665F),8 which were subjected to simulate and investigate a loss of interactions between patient-derived monoclonal antibodies and these classic exosite-3 epitope residues. As shown previously8 and here, II-1 and I-9 can no longer bind to GoF-ADAMTS13. The WT and GoF complexes were used in molecular dynamics simulations (Figure 1C; supplemental Video 1, available on the Blood Web site)16 to determine the most likely binding pose for II-1 and I-9 by means of binding free energy (BFE) calculation,17-19 and the lowest BFE (highest affinity) for the WT–antigen–autoantibody complex (compared with the GoF–antigen–autoantibody complex; Figure 1D). The selected complexes for I-9 and II-1 were further investigated at a residue level on the spacer domain, and a larger epitope landscape was predicted by our models. These predicted residues were mutated to an alanine. Alternatively, putative N-glycan attachment sequences were inserted in the loops where these residues are located (supplemental Table 1). These variants were produced in Chinese hamster ovary cells and quantified, and the binding of each variant was measured against II-1, I-9, and a cohort of previously characterized iTTP patients’ sera as previously described.10 The activity level of each variant was also measured with a FRETS-VWF7320 and VWF multimer assay.10 Selected variants were also measured with a flow assay. The presence of N-glycans was experimentally determined by mass spectrometry. Further details of the experimental procedures are available in the supplemental Material.
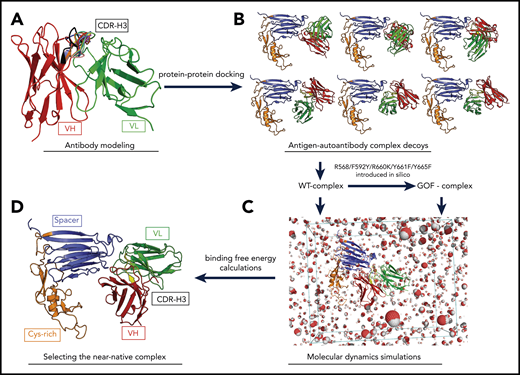
Structural bioinformatics pipeline to build an antigen-autoantibody complex. (A) Antibody modeling of the Fv fragments of the II-1 and I-9 autoantibodies. Heavy chain (VH; shown in red) and light chain (VL; shown in green) built and orientated with each other. Complementary determining region 3 of VH (CDR-H3; shown in multiple colors) was grafted at different conformations to the Fv model. (B) Docking results of ADAMTS13 antigen-autoantibody complex following the protein-protein docking step. (C) Molecular dynamics simulations for each WT- and GoF-complex models were run. (D) A likely complex between ADAMTS13 antigen and autoantibody was selected by BFE calculations. Cys, cysteine.
Structural bioinformatics pipeline to build an antigen-autoantibody complex. (A) Antibody modeling of the Fv fragments of the II-1 and I-9 autoantibodies. Heavy chain (VH; shown in red) and light chain (VL; shown in green) built and orientated with each other. Complementary determining region 3 of VH (CDR-H3; shown in multiple colors) was grafted at different conformations to the Fv model. (B) Docking results of ADAMTS13 antigen-autoantibody complex following the protein-protein docking step. (C) Molecular dynamics simulations for each WT- and GoF-complex models were run. (D) A likely complex between ADAMTS13 antigen and autoantibody was selected by BFE calculations. Cys, cysteine.
Results and discussion
Models of ADAMTS13 spacer and II-1/I-9 autoantibody complexes predict a larger epitope landscape
The antigen-autoantibody complex models predicted an expansion of the previously determined epitope area in autoantibody binding. The model of the spacer–I-9 complex predicted that residues R568, L591, F592, K608, M609, R636, L637, R660, Y661, Y665, and L668 together form a larger epitope. The model of spacer–II-I complex predicted involvement of the same residues as well as R639 (Figure 2A; supplemental Table 1). Unexpectedly, all of the novel single-alanine variants (Figure 2A, yellow) were still capable of binding to patient-derived monoclonal antibodies II-1 and I-9 (Figure 2B), whereas the single-alanine variants of classic epitope residues (Figure 2A, magenta) displayed a varying but much stronger reduction in binding. This may reflect a lower contribution of L591, K608, M609, R636, L637, and L668 to I-9 and II-1 binding compared with several of the core classic R568/F592/R660/Y661/Y665 residues, despite the predicted interactions (supplemental Table 1).
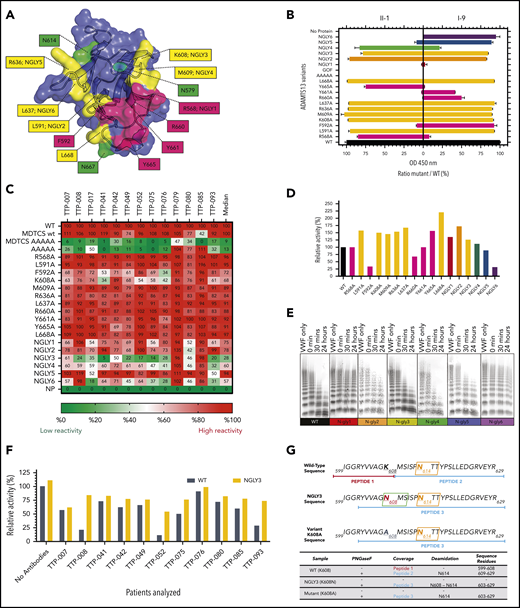
Measuring the autoantibody reactivity and proteolytic activity of novel ADAMTS13 variants. (A) ADAMTS13 epitope expansion based on modeling. Known epitope residues are depicted in magenta; predicted residues are depicted in yellow. Native N-glycan residues are highlighted in green. Artificial N-glycans were introduced at positions 568 (NGLY1), 591 (NGLY2), 608 (NGLY3), 609 (NGLY4), 636 (NGLY5), and 637 (NGLY6). (B) Reactivity of ADAMTS13 variants against II-1 and I-9 monoclonal autoantibodies. (C) Reactivity of ADAMTS13 variants against TTP patient sera in heat map format. Numbers represent relative binding compared with WT (in percentage; 100% is red, 50% is white, and 0% is green). (D) Relative activity of ADAMTS13 variants in FRETS-VWF73 assay (WT-ADAMTS13 is 100%). (E) Activity of NGLY-ADAMTS13 variants in VWF multimer assay. (F) Relative activity of ADAMTS13 variants measured with FRETS-VWF73 in the presence of TTP patient sera (WT-ADAMTS13 is 100%; all values compared with WT-ADAMTS13 without any patient antibodies). (G) Mass spectrometry reveals presence of an N-glycan at amino acid position 608 in NGLY3 (K608N). The presence of the N-glycan at this position is derived from the detection of a K608N containing deamidated peptide of NGLY3 upon treatment with PNGaseF (for further details see supplemental Materials). OD, optical density.
Measuring the autoantibody reactivity and proteolytic activity of novel ADAMTS13 variants. (A) ADAMTS13 epitope expansion based on modeling. Known epitope residues are depicted in magenta; predicted residues are depicted in yellow. Native N-glycan residues are highlighted in green. Artificial N-glycans were introduced at positions 568 (NGLY1), 591 (NGLY2), 608 (NGLY3), 609 (NGLY4), 636 (NGLY5), and 637 (NGLY6). (B) Reactivity of ADAMTS13 variants against II-1 and I-9 monoclonal autoantibodies. (C) Reactivity of ADAMTS13 variants against TTP patient sera in heat map format. Numbers represent relative binding compared with WT (in percentage; 100% is red, 50% is white, and 0% is green). (D) Relative activity of ADAMTS13 variants in FRETS-VWF73 assay (WT-ADAMTS13 is 100%). (E) Activity of NGLY-ADAMTS13 variants in VWF multimer assay. (F) Relative activity of ADAMTS13 variants measured with FRETS-VWF73 in the presence of TTP patient sera (WT-ADAMTS13 is 100%; all values compared with WT-ADAMTS13 without any patient antibodies). (G) Mass spectrometry reveals presence of an N-glycan at amino acid position 608 in NGLY3 (K608N). The presence of the N-glycan at this position is derived from the detection of a K608N containing deamidated peptide of NGLY3 upon treatment with PNGaseF (for further details see supplemental Materials). OD, optical density.
N-glycan variants of ADAMTS13 can resist against II-1 and I-9
Putative N-glycan motifs (supplemental Table 1) were inserted as a complementary approach to cause a steric hindrance on the spacer epitope for autoantibodies. As shown in Figure 2B, variants NGLY1, NGLY5, and NGLY6 were able to resist against autoantibody II-1, and only NGLY1 and NGLY4 were partly resistant against I-9. NGLY2 and NGLY3 did not show any resistance against II-1 and I-9.
Variant NGLY3 can resist against autoantibody repertoires of TTP patients
All above-mentioned variants of ADAMTS13 were tested against a cohort of iTTP patients’ sera (n = 13). As previously shown,10 single-alanine mutations did not significantly reduce binding against patient sera (Figure 2C). Only F592A and K608A were able to cause a mild reduction to ∼70% of WT-ADAMTS13 binding. In contrast, a reduced median binding (up to 50% to 55%) of patients’ autoantibodies to NGLY1, NGLY2, NGLY4, and NGLY6 was observed. Binding of patient-derived autoantibodies to NGLY5 was only mildly affected. Interestingly, variant NGLY3 showed the lowest median binding against the patient’s sera (28%), suggesting that insertion of an N-glycan at amino acid position 608 efficiently shields binding of autoantibodies to the spacer domain in 10 of 13 patients (<50%). In the rest of the patients, 2 out of 3 had C-terminal autoantibodies (TTP-079 and TTP-085) (Figure 2C).
Proteolytic activity of ADAMTS13 variants
Subsequently, we addressed whether the ADAMTS13 variants described above were still capable of cleaving VWF. Among all single-alanine variants, F592A and R660A were the only variants that resulted in reduced activity relative to WT-ADAMTS13 in FRETS-VWF73 (Figure 2D). Other exosite-3 variants (R568A and Y661A) retained an identical activity to WT-ADAMTS13. Variant Y665A, together with all other expanded-epitope single-alanine mutant variants (L591A, K608A, M609A, R636A, L637A and L668A), resulted in higher activity than WT-ADAMTS13 in FRETS-VWF73. The activity of single-alanine ADAMTS13 variants tested against recombinant VWF multimers in a static assay is in concordance with results obtained using FRETS-VWF73 (supplemental Figure 1).
With FRETS-VWF73, variants NGLY1-5 displayed similar or higher activity when compared with WT-ADAMTS13. Variant NGLY6 was significantly less active than WT-ADAMTS13 (Figure 2D). NGLY3 and NGLY4 were similarly active as WT-ADAMTS13 (Figure 2D-E). The ability of NGLY1, NGLY2, NGLY5, and NGLY6 to process VWF multimers was reduced, likely because of increased urea sensitivity (Figure 2E; supplemental Figure 2). In addition, NGLY3 and WT-ADAMTS13 had similar levels of proteolytic activity against VWF multimers under flow conditions (supplemental Figure 6). NGLY3 was subsequently tested for activity together with an excess of patient autoantibodies in a FRETS-VWF73 assay.10 Seven out of 11 patients (63%) inhibited the NGLY3-ADAMTS13 variant less when compared with the WT-ADAMTS13 (Figure 2F; supplemental Figure 4). The remaining 4 patients inhibited both the WT- and NGLY3-ADAMTS13 to a similar extent. The presence of an additional N-glycan on NGLY3-ADAMTS13 was confirmed in mass spectrometry by the detection of K608N deamidation upon deglycosylation (Figure 2G; supplemental Figure 5). We conclude that the significant reduction in reactivity with NGLY3-ADAMTS13 (K608N) against TTP patient sera is caused by the steric hindrance created by the N-glycan. Based on these properties, we suggest that the NGLY3-ADAMTS13 variant may potentially be used to rapidly restore the VWF-ADAMTS13 axis in patients suffering from acute iTTP.
Requests for data may be made by contacting the corresponding author, Jan Voorberg, at j.voorberg@sanquin.nl.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This project received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement 675746 (PROFILE) and from The Netherlands Ministry of Health (PPOC-18-022). B.E., N.A.G.G., K.K., J.H., K.V., A.V., P.C., C.R., G.A.F.N., A.M., and J.V. are members of the PROFILE Consortium.
Authorship
Contribution: B.E., N.A.G.G., G.A.F.N., and J.V. designed the study; K.K., A.M., and N.A.G.G. constructed and expressed ADAMTS13 variants; B.E., N.A.G.G., K.K., T.A., and P.K. performed experiments; B.E., J.H., G.A.F.N., C.R., and K.W. performed or supervised the computational modeling; K.V. provided antibodies 3H9, biotinylated 17G2, and 19H4 for quantitation of recombinant ADAMTS13; A.V. and P.C. provided patient samples; F.P.J.v.A. and M.v.d.B. performed mass spectrometry; B.E., N.A.G.G., and J.V. wrote the manuscript; and all authors critically revised the manuscript before submission.
Conflict-of-interest disclosure: B.E., N.A.G.G., and J.V. are inventors on a patent application regarding autoantibody-resistant ADAMTS13 variants. P.C. is a member of the clinical advisory board for Shire-Takeda. A.V. and K.V. are members of the scientific advisory boards of Ablynx-Sanofi and Shire-Takeda. The remaining authors declare no competing financial interests.
The current affiliation for B.E. is Division of Biochemistry, Netherlands Cancer Institute, Amsterdam, The Netherlands.
Correspondence: Jan Voorberg, Department of Molecular and Cellular Hemostasis, Sanquin-Academic Medical Center Landsteiner Laboratory, Amsterdam, The Netherlands; e-mail: j.voorberg@sanquin.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal