

In this issue of Blood, 1 report a higher frequency of reduced plasminogen-activating capacity by peripheral blood mononuclear cells (PBMCs) from patients with thrombosis and a positive family history, but not a known inherited thrombophilia, compared with either patients without a family history of thrombosis or patients with a family history accompanied by an inherited thrombophilia (see figure). Deficient cell surface plasmin generation by PBMCs, used in this study as a surrogate for endothelial cells, was caused by decreased expression of annexin A2. This report extends previous studies, primarily in animal models, suggesting an important role for annexin A2–mediated plasmin generation on the vascular wall in the maintenance of blood fluidity and suggests that deficient annexin A2 expression may represent a novel variant of thrombophilia that is independent of altered levels and/or mutations in circulating coagulation proteins or their inhibitors.
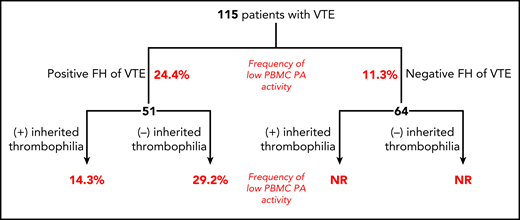
Distribution of 115 patients with VTE by family history and absence or presence of known inherited thrombophilia. Numbers in red depict percentages of patients with PBMCs expressing PA activity in lowest 10th percentile of the study population. FH, family history; NR, not reported.
Distribution of 115 patients with VTE by family history and absence or presence of known inherited thrombophilia. Numbers in red depict percentages of patients with PBMCs expressing PA activity in lowest 10th percentile of the study population. FH, family history; NR, not reported.
Annexin A2 is 1 of a family of 12 annexins and is highly conserved among mammalian species. It has been associated with a remarkable array of biologic functions, most of which depend on its membrane binding and organizing properties. Annexin A2 is expressed in many cell types, including endothelial cells, monocytes, macrophages, trophoblasts, and tumor cells, among others.2 In endothelial cells, the majority of annexin A2 is present in the cytoplasm, existing largely as a 36-kDa monomer, with only about 4% translocated to the external plasma membrane in the form of a heterotetramer with S100A10 (p11). The mechanism by which annexin A2 is translocated to the cell surface is not well understood; translocation is stimulated by thrombin, hypoxia, and other cell activators or stressors and is dependent on phosphorylation of Y23 by a pp60src-like kinase. Translocation also requires expression of p11, which is stabilized by binding to annexin A2.3
By binding both tissue plasminogen activator (t-PA) and plasminogen, the annexin A2•p11 heterotetramer mediates activation of plasminogen by t-PA.4 t-PA binds to an LCKLSL sequence in the N-terminal tail of annexin A2, whereas plasminogen binds to a C-terminal lysine (K308) of annexin A2, thought to be generated by partial annexin A2 proteolysis5 and/or the C-terminal lysine of p11.6 Through binding both reactants, annexin A2 lowers the KM and increases the catalytic efficiency of t-PA–mediated plasminogen activation by ∼60-fold4 ; in the context of endothelial cells, this leads to increased vascular wall fibrinolytic activity.
There are several examples of the in vivo relevance of annexin A2–mediated plasminogen activation on vascular function. Annexin A2–deficient mice accumulate fibrin in multiple intra- and extravascular sites, and develop occlusive thrombi in the FeCl3 carotid thrombosis model more frequently than wild-type littermates.7 Infusion of annexin A2 along with t-PA in rat stroke models reduces thrombus and infarct size, and a subgroup of patients with thrombotic antiphospholipid syndrome have antibodies reactive with annexin A2, which inhibit annexin A2–mediated plasminogen activation on endothelial cells.2 Recent studies have also implicated reduced maternal annexin A2 expression in the pathogenesis of preeclampsia. In contrast, excessive annexin A2 expression is associated with hyperfibrinolysis and bleeding in patients with promyelocytic leukemia because of robust expression of annexin A2 on the circulating leukemic cells.8 Recent clinical studies also suggest a positive correlation between annexin A2 expression and the aggressiveness of malignant tumors.
Despite the enthusiasm generated by the discovery of the many causes of inherited thrombophilia, an inherited thrombophilia cannot be identified in a substantial proportion of patients who present with thrombosis, even those with a positive family history; in the current study, 51 of 115 patients had a family history of venous thromboembolism (VTE), with an inherited thrombophilia identified in only 19 of these 51 individuals. When the capacity of PBMCs from these 19 individuals to support the activation of plasminogen was measured, 14.3% fell into the bottom 10th percentile of the study population; in comparison, the frequency of low plasminogen activation by PBMCs from the 32 patients with a positive family history but no identified inherited thrombophilia was more than twice as high (29.2%). Deficient plasminogen activation was correlated with a 70% decrease in annexin A2 protein and 30% decrease in mRNA; a 5 single nucleotide polymorphism cluster in the annexin A2 gene1 was more common in patients with VTE and low annexin A2 protein levels but did not correlate with annexin A2 mRNA levels. These studies provide compelling data supporting a role for decreased annexin A2 expression as a potential underlying cause of inherited thrombophilia.
These findings suggest additional questions and directions for future studies. First, it would be of interest to determine the incidence of deficient plasminogen activation in VTE patients without a family history, either with or without an inherited thrombophilia. Second, directly assessing annexin A2 expression and fibrinolytic activity in blood origin endothelial cells would be a fascinating, although challenging study. Third, defining the ability of deficient cell surface plasminogen activation to prospectively predict the risk of recurrent VTE would be clinically valuable; however, a more streamlined assay, perhaps flow cytometric assessment of PBMC cell surface annexin A2 expression, might need to be developed to provide a more practical clinical assay. Finally, the mechanism(s) underlying reduced annexin A2 expression, apparently on an inherited basis, remains incompletely defined and requires further study.
This study is among the first to suggest one of perhaps many yet to be discovered abnormalities in vascular wall function as a cause of inherited vascular thrombophilia and opens the door to broader understanding and investigation of this important concept.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
