

Key Points
Those asymptomatic at the time of HSCT experienced normal to near-normal development in all developmental domains.
Those with disease onset at >12 months of age had better outcomes than onset ≤12 months, supporting a reclassification of late-infantile Krabbe disease.

Abstract
Krabbe disease is a rare neurodegenerative disorder caused by a deficiency in galactocerebrosidase. The only effective treatment is hematopoietic stem cell transplantation (HSCT). Approximately 85% of Krabbe disease cases are the infantile subtypes, among which ∼20% are late infantile. Prior studies have demonstrated that HSCT is effective for early-infantile patients (0-6 months of age) who undergo transplantation while asymptomatic, compared with those receiving transplants while symptomatic. However, no studies evaluated the efficacy of HSCT for late-infantile patients (6-36 months). In this prospective, longitudinal study, patients were evaluated at a single site according to a standardized protocol. Survival analysis was performed using the Kaplan-Meier method. Differences between groups were estimated using mixed regression models to account for within-person repeated measures. Nineteen late-infantile patients underwent HSCT (March 1997 to January 2020). Compared with untreated patients, transplant recipients had a longer survival probability and improved cognitive and language function. Gross and fine motor development were most affected, with variable results. Asymptomatic patients benefitted the most from transplantation, with normal to near-normal development in all domains and some gross motor delays. Among symptomatic patients, those with disease onset at >12 months of age had better cognitive outcomes than untreated patients. Those with disease onset at ≤12 months were comparable to untreated patients. We found that HSCT prolonged the lifespan and improved the functional abilities of late-infantile patients with Krabbe disease, particularly those who underwent transplantation before onset of symptoms. In addition, our findings support prior literature that reclassifies late-infantile Krabbe disease to be symptom onset at 12 to 36 months of age.
Introduction
Krabbe disease is an autosomal recessive disorder resulting from a deficiency of galactocerebrosidase (GALC).1-3 GALC deficiency leads to an accumulation of galactocerebroside and its parent cytotoxic compound, galactosylsphingosine, both of which are toxic to the nervous system. Accumulation of galactosylsphingosine elicits the death of oligodendrocytes and myelin arrest, leading to progressive demyelination.3
Krabbe disease is divided into subtypes based on age of symptom onset (early infantile, late infantile, juvenile, and adult). Diagnosis is made by measuring GALC activity in fibroblasts or white cells and is confirmed by mutation analysis. Prior studies showed that siblings of individuals with infantile phenotypes have similar age at disease onset, as well as disease trajectory, in contrast to those with juvenile and adult onset, in which variability may be seen.4
Traditionally, early-infantile Krabbe disease was thought to have onset from 0 to 6 months and late-infantile from 6 to 36 months. However, Bascou et al found that patients with onset from 6 to 12 months had disease presentation resembling that observed in early-infantile patients.1 Thus, early infantile was recently reclassified as symptom onset from 0 to 12 months and late-infantile from 12 to 36 months.
Most patients (∼85% of Krabbe disease cases) have the infantile phenotype. The early infantile phenotype (0-12 months) is the most aggressive form of the disease, with an incidence rate of ∼1 to 2 in 100 000.1-6 Comparatively, the late-infantile onset occurs between 13 and 36 months. This form has an incidence of ∼20% and presents with delay in motor milestones, psychomotor regression, ataxia, irritability, and loss of vision.1,4,6,7
To date, hematopoietic stem cell transplantation (HSCT) is the only effective treatment for Krabbe disease.8-13 Previous studies investigating HSCT in early-infantile patients found that when transplantation was performed before onset of symptoms, the patient experienced significant improvements in both lifespan and functional ability.8,9
Although there have been long-term studies of patients with onset before 6 months of age, there is a gap in the literature regarding the efficacy of HSCT in late-infantile patients, with the exception of a few case studies. Given the increasing number of states that are adding Krabbe disease to the newborn screening (NBS) panels, there is a need for studies investigating the outcomes of HSCT in patients with late-onset Krabbe disease. The purpose of this study was to describe the long-term transplantation outcomes of children with Krabbe disease who have onset between 6 and 36 months of age.
Methods
Study population and study design
This study included children with late-infantile Krabbe disease referred to the Program for the Study of Neurodevelopment in Rare Disorders (NDRD) between 1997 and 2020, who underwent HSCT. Because this project began before the reclassification, the late-infantile patients of this study follow the traditional classification of having symptom onset between 6 and 36 months. The baseline data before transplantation are included in the natural history reported in Bascou et al.1
Diagnosis was confirmed by measuring GALC activity. Genetic data were collected when available. Because mutation analysis was not a standard for diagnosis until ∼2009, we were unable to obtain a genotype for some patients.
At each visit, patients were evaluated at a single site by neurodevelopmental pediatricians, speech pathologists, audiologists, physical therapists, and psychometricians with experience in evaluating children with neurodegenerative diseases. A comprehensive, standardized, and prospectively designed protocol for long-term neurodevelopmental assessment of lysosomal storage diseases was used to evaluate signs and symptoms of disease, growth, motor function, cognitive and adaptive skills, physical and neurological changes, sensory function, and language skills.14 Other tests included were nerve conduction velocity (NCV), magnetic resonance imaging (MRI), electroencephalogram (EEG), visual evoked potential (VEP), and auditory brainstem response (ABR).
For analysis, patients were classified as asymptomatic or symptomatic. Before transplantation, all patients underwent a thorough examination to assess for initial signs and symptoms of late-infantile Krabbe disease: irritability, spasticity, loss of acquired developmental milestones, abnormal gait, and abnormal muscle tone.1 In addition, patients were evaluated for signs and symptoms commonly found in Krabbe disease (supplemental Table 1, available on the Blood Web site). Symptomatic patients were those who underwent transplantation after onset of initial signs and symptoms. Asymptomatic patients were those who had the procedure before onset of initial symptoms and who had an affected older sibling, allowing for early diagnosis. Siblings of individuals with infantile phenotypes have similar onsets and disease trajectories.4
Comparisons were made between this study’s cohort of late-infantile transplant recipients with Krabbe disease and untreated late-infantile patients. The untreated patient data are from the natural history conducted by our group, reported by Bascou et al.1
The study was approved by the institutional review boards of the University of North Carolina (IRB-08-0237) and the University of Pittsburgh (IRB-PRO11050036). Research was conducted in accordance with the Declaration of Helsinki.
Transplantation procedure
Patients underwent either umbilical cord blood transplantation (UCBT) or bone marrow transplantation (BMT). Some patients who underwent UCBT received full conditioning, and some received a reduced-intensity conditioning (RIC) regimen designed for nonmalignant disorders (Pitt-RIC). The full conditioning was based on busulfan and cyclophosphamide. The details of the Pitt-RIC, which is not busulfan-based, can be found in Vander Lugt et al.15
GALC, psychosine, and CSF analysis
All GALC activity measurements were conducted at the Lysosomal Diseases Testing Laboratory at Thomas Jefferson University. GALC testing at the laboratory is based on the percentage of the daily mean. The level of all lysosomal enzymes is taken into account to control for variability in reagents used in white cell lysates of leukocytes or fibroblasts. Diagnosis was established by David Wenger, director of the laboratory.
The natural history of late-infantile Krabbe disease indicated that patients have GALC of 0 to 0.29 nmol per hour per milligram protein.1 We considered patients with levels <0.5 nmol per hour per milligram protein to be potentially at risk.
All psychosine measurements were made from dried blood spots and analyzed in Michael Gelb’s laboratory at the University of Washington.16 At the time of measurement, normal ranges of psychosine were not yet established.
Cerebrospinal fluid (CSF) was obtained by lumbar puncture while the patient was under anesthesia for MRI or local anesthesia. The total CSF protein level was determined by tandem mass spectrometry.
Neurodevelopmental analysis
To assess cognitive and language function, the Early Learning Composite of the Mullen Scales of Early Learning was used. When patients were >4 years of age developmentally, the General Conceptual Ability composite score from the Differential Ability Scales was used, including the Preschool, Early Years, and School-Age batteries. The composite score from the Cognitive Adaptive Test/Clinical Linguistic and Auditory Milestone Scale was used in the early years of the study.17-20
To assess gross motor function, patients functioning at <5 years of age were evaluated with either the Peabody Developmental Motor Scales or the Gross Motor subtest of the Mullen Scales of Early Learning. Patients’ functioning at >5 years of age was evaluated with the Bruininks-Oseretsky Test of Motor Proficiency.17,21-23
To assess adaptive skills and development, the Vineland Adaptive Behavior Scales or the Scales of Independent Behavior were used.24,25
Neuroradiologic and neurophysiologic analysis
MRI scans (sagittal T1 fluid-attenuated inversion recovery and T2 sampling perfection with sampling perfection with application-optimized contrasts using different flip angle evolutions, and axial dual-echo T2 and proton density–weighted brain MRI) were obtained by using a 3-Tesla scanner. Scans were interpreted by an experienced neuroradiologist and evaluated for any abnormalities. Over the 21-year period of this study, there have been 3 neuroradiologists. Examiners were not blinded, because the imaging was performed for clinical assessments. To determine signs of disease progression, the following were assessed in the scans: signal intensity, involvement of additional regions that were unaffected in previous scans, and severity of atrophy. To determine whether the patient had improved, stabilized, or worsened in disease progression, the neuroradiologist compared the current scans with the prior ones and considered the changes expected in normal aging.
Flash VEPs were evaluated as normal if the P100 wave was present and as abnormal if the P100 wave was absent.
ABRs were considered abnormal if wave 1 to 5 interpeak latencies were prolonged or any of the obligate waveforms (1, 3, or 5) was absent.
NCV motor responses were measured in the peroneal, tibial, and ulnar nerves. Sensory responses were measured in the sural and median nerves. NCV results were considered abnormal if they showed prolongation of distal and F-wave latencies, low amplitude, or no evoked response.
Statistical analysis
For patients lost to follow-up, The Social Security Death Index was queried to search for any deaths. Internet searches were also conducted to find mention of the patient’s death. Survival was estimated by the Kaplan-Meier method, using the cutoff date of 1 January 2020. Clinical growth charts were created based on the published Center for Disease Control growth charts.26 Developmental growth charts were created by plotting the patient’s age-equivalent score against actual age. Age-equivalent scores are ideal for longitudinal analysis in neurodegenerative disorders, as they can be used to ascertain whether a child is gaining or losing skills over time. The group mean curve was estimated based on data from the World Health Organization.
Mixed regression models were fitted to test for group differences with the age-equivalent score as the dependent variable, and group, age, and the group by age interaction as independent variables. One-sided postestimation Student t tests were used to test for differences between the groups’ development over time (ie, interaction of group × age).
Results
Patient characteristics
The 19 patients in this study were 15 boys and 4 girls (16 White, 2 Black, 1 Black and Hispanic) who underwent HSCT. Five patients were asymptomatic and 14 were symptomatic. Mean pretransplantation GALC levels were 0.027 nmol per hour per milligram protein (range, 0.0-0.13; Table 1). Pretransplantation CSF protein levels ranged from 50 to 296 mg/dL (normal, <40; Table 2).
Summary of patients’ GALC mutations and activity levels before and after transplantation
| Patient | GALC activity(nmol/h/mg protein) | GALC mutation | ||||
|---|---|---|---|---|---|---|
| Pre-HSCT | Post-HSCT | Allele 1 | Allele 2 | |||
| DNA | Protein | DNA | Protein | |||
| Asymptomatic | ||||||
| 1 | 0.05 | 6.1 | — | — | — | — |
| 2 | 0 | 2.4 | — | — | — | — |
| 3 | 0.04 | 1.8 | — | — | — | — |
| 4 | 0.08 | 2.4 | — | — | — | — |
| 5 | 0 | 3.7 | c.349A>G | p.Met117Val | c.908C>T | p.Ser303Phe |
| c.1685T>C* | p.Ile562Thr* | c.1685T>C* | p.Ile562Thr* | |||
| Symptomatic | ||||||
| 6 | 0.01 | 4.5 | — | — | — | — |
| 7 | 0 | 3.1 | — | — | — | — |
| 8 | 0 | 2.4 | — | — | — | — |
| 9 | 0.05 | 4.1 | — | — | — | — |
| 10 | 0.13 | 4.1 | — | — | — | — |
| 11 | 0 | 2.8 | — | — | — | — |
| 12 | 0 | 0.4 | — | — | — | — |
| 13 | 0.07 | 1 | c.349A>G | p.Met117Val | c.909-10A>G | — |
| c.1685T>C* | p.Ile562Thr* | c.1685T>C* | p.Ile562Thr* | |||
| 14 | 0.06 | 1.3 | c.379C>T | p.Arg127Xaa20 | c.850G>A | p.Gly284Ser |
| c.1685T>C* | p.Ile562Thr* | c.1685T>C* | p.Ile562Thr* | |||
| 15 | 0 | 2.8 | c.674C>A | p.Ala225Glu21 | c.850G>A | p.Gly284Ser |
| — | — | c.1685T>C* | p.Ile562Thr* | |||
| 16 | 0 | 0.35 | c.916G>A | p.Ala306Thr | c.30Kb del | — |
| 17 | 0.08 | 1.3 | — | — | — | — |
| 18 | 0 | 0.32 | — | — | — | — |
| 19 | 0.094 | 1.1 | c.857G>A | p.Gly286Asp | c.1161+6532 | polyA+9kbdel |
| Patient | GALC activity(nmol/h/mg protein) | GALC mutation | ||||
|---|---|---|---|---|---|---|
| Pre-HSCT | Post-HSCT | Allele 1 | Allele 2 | |||
| DNA | Protein | DNA | Protein | |||
| Asymptomatic | ||||||
| 1 | 0.05 | 6.1 | — | — | — | — |
| 2 | 0 | 2.4 | — | — | — | — |
| 3 | 0.04 | 1.8 | — | — | — | — |
| 4 | 0.08 | 2.4 | — | — | — | — |
| 5 | 0 | 3.7 | c.349A>G | p.Met117Val | c.908C>T | p.Ser303Phe |
| c.1685T>C* | p.Ile562Thr* | c.1685T>C* | p.Ile562Thr* | |||
| Symptomatic | ||||||
| 6 | 0.01 | 4.5 | — | — | — | — |
| 7 | 0 | 3.1 | — | — | — | — |
| 8 | 0 | 2.4 | — | — | — | — |
| 9 | 0.05 | 4.1 | — | — | — | — |
| 10 | 0.13 | 4.1 | — | — | — | — |
| 11 | 0 | 2.8 | — | — | — | — |
| 12 | 0 | 0.4 | — | — | — | — |
| 13 | 0.07 | 1 | c.349A>G | p.Met117Val | c.909-10A>G | — |
| c.1685T>C* | p.Ile562Thr* | c.1685T>C* | p.Ile562Thr* | |||
| 14 | 0.06 | 1.3 | c.379C>T | p.Arg127Xaa20 | c.850G>A | p.Gly284Ser |
| c.1685T>C* | p.Ile562Thr* | c.1685T>C* | p.Ile562Thr* | |||
| 15 | 0 | 2.8 | c.674C>A | p.Ala225Glu21 | c.850G>A | p.Gly284Ser |
| — | — | c.1685T>C* | p.Ile562Thr* | |||
| 16 | 0 | 0.35 | c.916G>A | p.Ala306Thr | c.30Kb del | — |
| 17 | 0.08 | 1.3 | — | — | — | — |
| 18 | 0 | 0.32 | — | — | — | — |
| 19 | 0.094 | 1.1 | c.857G>A | p.Gly286Asp | c.1161+6532 | polyA+9kbdel |
Variants are reported using Human Genome Variation Society nomenclature27 reference sequences NP_000144.2 (protein) and NM_000153.3 (cDNA nucleotide). The patient numbers align with those in supplemental Table 1.
−, no information available.
A polymorphism that has been reported to have no pathogenicity when present without another accompanying pathogenic variant.28
CSF protein levels in all patients before and after transplantation
| Patient | CSF (mg/dL) | |
|---|---|---|
| Pre-HSCT | Post-HSCT | |
| Asymptomatic | ||
| 1 | NA | NA |
| 2 | NA | 29 |
| 3 | NA | 104 |
| 4 | 55 | 129 |
| 5 | NA | 18 |
| Symptomatic | ||
| 6 | NA | 39 |
| 7 | 296 | NA |
| 8 | 222 | 140 |
| 9 | 83 | 174 |
| 10 | NA | 191 |
| 11 | 214 | NA |
| 12 | 95 | 127 |
| 13 | 90 | NA |
| 14 | 117 | 114 |
| 15 | 174 | 148 |
| 16 | 146 | 90 |
| 17 | 50 | 17 |
| 18 | 204 | 205 |
| 19 | 78 | NA |
| Patient | CSF (mg/dL) | |
|---|---|---|
| Pre-HSCT | Post-HSCT | |
| Asymptomatic | ||
| 1 | NA | NA |
| 2 | NA | 29 |
| 3 | NA | 104 |
| 4 | 55 | 129 |
| 5 | NA | 18 |
| Symptomatic | ||
| 6 | NA | 39 |
| 7 | 296 | NA |
| 8 | 222 | 140 |
| 9 | 83 | 174 |
| 10 | NA | 191 |
| 11 | 214 | NA |
| 12 | 95 | 127 |
| 13 | 90 | NA |
| 14 | 117 | 114 |
| 15 | 174 | 148 |
| 16 | 146 | 90 |
| 17 | 50 | 17 |
| 18 | 204 | 205 |
| 19 | 78 | NA |
Seventeen patients underwent UCBT, and 2 underwent BMT (1 related allogenic from a sibling; 1 from an unrelated donor). Five patients who underwent UCBT received the Pitt-RIC regimen.
Survival and engraftment
At the time of publication, 14 patients were alive and 5 were deceased. Two patients died of peritransplantation complications, 1 patient died after losing his graft (BMT; died 2.67 years after HSCT), 1 patient died of disease progression (5.6 years after HSCT), and 1 patient died of an infection (chicken pox; died 1.2 years after HSCT). The estimated probability of survival up to 25 years of age for transplant recipients is 0.725 (95% confidence interval [CI], 0.54-0.97). For untreated patients, the median mortality was 6.72 years, with the probability of living to 11 years at 0.13 (95% CI, 0.02-0.67; Figure 1).
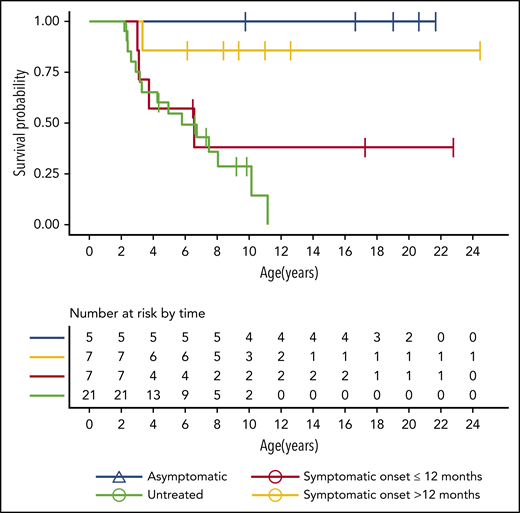
All patients were engrafted but 1 did not maintain the graft (the patient who underwent unrelated BMT). All patients who underwent UCBT with Pitt-RIC maintained the graft and survived.
All patients had normal GALC enzyme activity levels after transplantation, with 3 at the lower end of normal limits (0.32, 0.35, and 0.4; Table 1). Posttransplantation GALC levels were measured 1 month to 9.5 years after transplantation (mean, 34.4 months).
Among patients with data available, all but 1 (the patient who lost his graft) had psychosine levels that declined after HSCT (Figure 2).
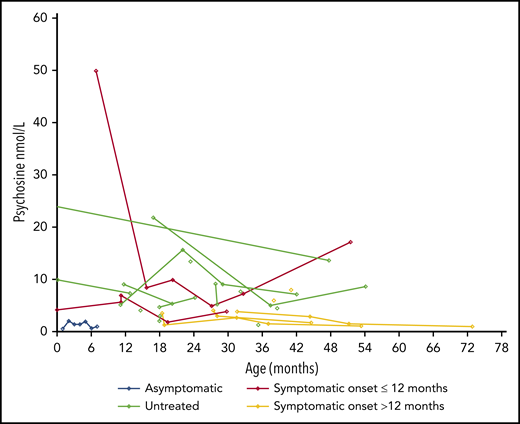
Psychosine levels at various ages in the patients with data available. Each line represents a single patient, and each circle indicates an individual measurement.
Psychosine levels at various ages in the patients with data available. Each line represents a single patient, and each circle indicates an individual measurement.
Growth and nutrition
Among the 17 patients with available data, 13 were below the 50th percentile for weight and height. All had head circumferences within 2 standard deviations from the mean, except for 1 with microcephaly (Figure 3).
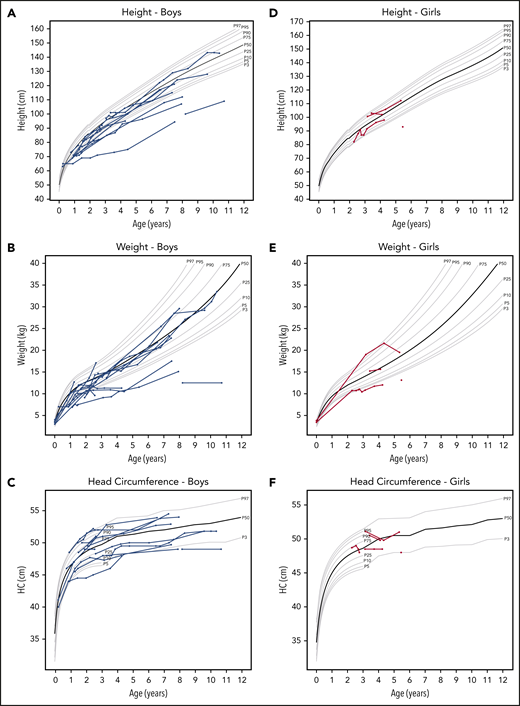
Height, weight, and head circumference of the patients. Each line represents a patient, and each circle indicates an individual measurement. The gray lines show the standard growth curves for the 3rd, 5th, 10th, 25th, 50th, 75th, 90th, 95th, and 99th percentiles.18 In the graphs on the left (A-C), blue lines indicate boys, and in the graphs on the right (D-F), red lines indicate the girls.
Height, weight, and head circumference of the patients. Each line represents a patient, and each circle indicates an individual measurement. The gray lines show the standard growth curves for the 3rd, 5th, 10th, 25th, 50th, 75th, 90th, 95th, and 99th percentiles.18 In the graphs on the left (A-C), blue lines indicate boys, and in the graphs on the right (D-F), red lines indicate the girls.
Eighteen patients had nutrition data. At the most recent evaluation, 14 patients (78%) were able to eat by mouth, with 1 using a gastrostomy tube for medications and 2 using a gastrostomy tube for nutritional supplementation. The remaining patients (22%) exclusively used a gastrostomy tube, as they could not take in nutrition orally.
Neurodevelopmental outcomes
Overall development
For all domains except gross motor development, asymptomatic children developed normally (within the fifth to 95th percentiles) or near normally. Symptomatic patients were divided into those with symptom onset at >12 months of age and those with onset at ≤12 months. For symptomatic patients with onset at >12 months, development was delayed (below the fifth percentile) but all gained skills over time. Symptomatic patients with onset at ≤12 months performed similar to untreated patients (Figure 4).
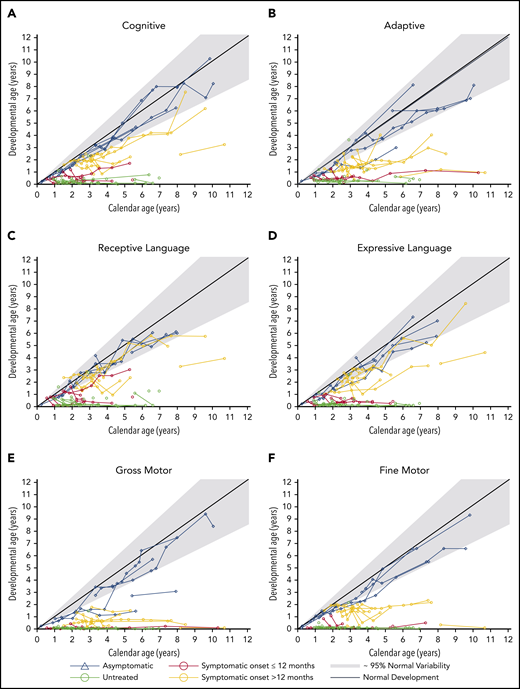
Neurodevelopmental outcomes of transplant recipients. Neurodevelopmental domains shown are cognitive development (A), adaptive behavior (B), receptive language (C), expressive language (D), gross motor (E), and fine motor (F). The developmental ages were determined by age-equivalent scores from tests. Each line represents a patient and each circle indicates an individual measurement. Gray lines and areas indicate normal development at the 95th, 50th, and fifth percentiles.
Neurodevelopmental outcomes of transplant recipients. Neurodevelopmental domains shown are cognitive development (A), adaptive behavior (B), receptive language (C), expressive language (D), gross motor (E), and fine motor (F). The developmental ages were determined by age-equivalent scores from tests. Each line represents a patient and each circle indicates an individual measurement. Gray lines and areas indicate normal development at the 95th, 50th, and fifth percentiles.
Cognitive development
All asymptomatic children developed cognitive skills at a normal rate.
All symptomatic patients with onset at >12 months continued to gain skills. Half of the patients had a near-normal development rate, whereas the other half had delayed development.
Among symptomatic patients with onset at ≤12 months, only 1 continued to gain skills, albeit at a delayed rate. All other patients reached a developmental plateau, with skills corresponding to a 6-month-old (Figure 4A).
Adaptive behavior
Adaptive behavior is defined as the ability of children to take care of themselves independently within their community. All but 1 asymptomatic patient had normal development. The asymptomatic patient with delayed adaptive development also had delayed gross motor development.
Among symptomatic patients with onset at >12 months, most had delayed development. Only 2 patients plateaued at the developmental ages of 1.5 and 1 year old.
All symptomatic patients with onset at ≤12 months regressed and plateaued with skills below 1 year of age (Figure 4B).
Four (24%) asymptomatic children toileted independently, and 1 needed assistance. Three symptomatic patients needed assistance (23%), and 5 (38%) could not toilet independently. The remaining symptomatic patients were too young to toilet independently.
Receptive language development
All asymptomatic children had normal receptive language development.
Among symptomatic patients with onset at >12 months, 2 developed normally; 1 developed, albeit at a delayed rate; and the remaining 3 plateaued in development (developmental ages of 1.5 and 2 years). Among this symptomatic group, the 2 children whose language skills developed normally also showed the greatest gains in cognitive skills.
Except for 1 patient with near-normal development, all symptomatic patients with onset at ≤12 months plateaued with skills at or below 1 year of age (Figure 4C).
Expressive language development
All asymptomatic children had normal expressive language development.
All but 1 symptomatic patient with onset at >12 months continued gaining skills, among which 3 patients developed at a normal rate. Only 1 patient plateaued (developmental age of 2 years).
All symptomatic patients with onset at ≤12 months plateaued at the developmental level of a 5-month-old (Figure 4D).
Gross motor development
Three asymptomatic patients had normal gross motor development, 1 had delayed development, and 1 had normal development until 2.5 years of age, after which he plateaued at a developmental age of 1.5 years. This patient was noted to have generalized weakness from steroid myopathy. All asymptomatic children were able to walk independently.
All symptomatic patients experienced a plateau or regression of gross motor skills. Overall, patients with onset at >12 months plateaued with skills at a higher developmental age than those with onset at ≤12 months (Figure 4E). Only 2 symptomatic patients (onset at >12 months) were able to walk, with 1 needing an assistive device. All symptomatic patients with onset at ≤12 months were unable to walk.
Fine motor development
All asymptomatic patients had normal to near-normal fine motor development.
Similar to the trends seen in gross motor development, all symptomatic patients plateaued in fine motor development, but patients with onset at >12 months plateaued at a higher developmental age. Patients with onset at >12 months plateaued at ∼1.5 to 2 years of age whereas patients with onset at ≤12 months had minimal skills remaining (Figure 4F).
Group comparisons of longitudinal development of cognitive and gross motor abilities
Cognitive skills
Results from the mixed regression models indicated that asymptomatic children had normal cognitive development, with a slope of 0.96 vs 1.0 (slope observed for typically developing children; Table 3). The asymptomatic children’s rate of cognitive development was significantly higher than that of both symptomatic groups (>12 months: βdiff = 0.63, P < .001; ≤12 months: βdiff = 0.86, P < .001) and the untreated group (βdiff = 0.92; P < .001; Table 4).
Slope in years of the patient groups for cognitive and gross motor development
| Group | Cognitive development | Gross motor development | ||||
|---|---|---|---|---|---|---|
| β | SE | P | β | SE | P | |
| Asymptomatic | 0.96 | 0.09 | <.001 | 0.69 | 0.09 | <.001 |
| Symptomatic >12 mo | 0.32 | 0.08 | .003 | 0.01 | 0.08 | .87 |
| Symptomatic ≤12 mo | 0.10 | 0.11 | .63 | 0.02 | 0.12 | .87 |
| Untreated | 0.04 | 0.08 | .63 | 0.03 | 0.07 | .64 |
| Group | Cognitive development | Gross motor development | ||||
|---|---|---|---|---|---|---|
| β | SE | P | β | SE | P | |
| Asymptomatic | 0.96 | 0.09 | <.001 | 0.69 | 0.09 | <.001 |
| Symptomatic >12 mo | 0.32 | 0.08 | .003 | 0.01 | 0.08 | .87 |
| Symptomatic ≤12 mo | 0.10 | 0.11 | .63 | 0.02 | 0.12 | .87 |
| Untreated | 0.04 | 0.08 | .63 | 0.03 | 0.07 | .64 |
Differences in slope, in years, between the indicated groups for cognitive and gross motor development
| Groups | Cognitive development | Gross motor development | ||
|---|---|---|---|---|
| βdiff | P | βdiff | P | |
| Asymptomatic vs untreated | 0.92 | <.001 | 0.69 | <.001 |
| Asymptomatic vs symptomatic >12 mo | 0.63 | <.001 | 0.67 | <.001 |
| Asymptomatic vs symptomatic ≤12 | 0.86 | <.001 | 0.67 | <.001 |
| Symptomatic >12 mo vs untreated | 0.29 | .016 | −0.02 | .85 |
| Symptomatic ≤12 vs untreated | 0.06 | .65 | −0.01 | .92 |
| Symptomatic >12 mo vs symptomatic ≤12 | 0.23 | .11 | −0.01 | .97 |
| Groups | Cognitive development | Gross motor development | ||
|---|---|---|---|---|
| βdiff | P | βdiff | P | |
| Asymptomatic vs untreated | 0.92 | <.001 | 0.69 | <.001 |
| Asymptomatic vs symptomatic >12 mo | 0.63 | <.001 | 0.67 | <.001 |
| Asymptomatic vs symptomatic ≤12 | 0.86 | <.001 | 0.67 | <.001 |
| Symptomatic >12 mo vs untreated | 0.29 | .016 | −0.02 | .85 |
| Symptomatic ≤12 vs untreated | 0.06 | .65 | −0.01 | .92 |
| Symptomatic >12 mo vs symptomatic ≤12 | 0.23 | .11 | −0.01 | .97 |
The symptomatic >12-month cohort developed at a much slower rate than the asymptomatic cohort (βdiff = 0.63; P = .12) but performed better than the untreated patients, who did not gain any skills (βdiff = 0.29; P = .016). Symptomatic ≤12-month patients performed the same as untreated patients (βdiff = 0.06; P = .65).
Gross motor abilities
Asymptomatic children had abnormal gross motor development (slope = 0.69; SE = 0.09) but developed significantly better than both symptomatic groups (>12 month: βdiff = 0.67; P < .001; ≤12 month: βdiff = 0.67; P < .001) and the untreated group (βdiff = .69; P < .001). Neither the symptomatic groups nor the untreated group gained any significant skills (all P > .60; Table 4).
Neuroradiologic and neurophysiologic testing
MRI
Of 18 patients, 12 (67%) had stable or improved findings after transplantation.
Among the asymptomatic children, all stabilized (n = 3; 60%) or improved (n = 2; 40%; Table 6). Common brain regions with white matter abnormalities at the most recent examination were the periventricular (80%; n = 4; Table 5) and the centrum semiovale (n = 2; 40%; Table 5).
Proportion of patients with an abnormality in the specified brain region as determined by MRI results
| Brain region | Pre-HSCT, n (%) | Post-HSCT, n (%) |
|---|---|---|
| Asymptomatic (n = 5) | ||
| Brainstem | 2 (40) | 0 |
| Corpus callosum | 1 (20) | 0 |
| Corona radiata | 0 | 0 |
| Centrum semiovale | 1 (20) | 2 (40) |
| Corticospinal tracts | 0 | 0 |
| Periventricular | 0 | 4 (80) |
| Subcortical | 1 (20) | 1 (20) |
| U-fibers | 0 | 0 |
| Dentate nucleus | 1 (20) | 0 |
| Symptomatic (n = 13) | ||
| Brainstem | 5 (38) | 6 (46) |
| Corpus callosum | 6 (46) | 5 (38) |
| Corona radiata | 4 (31) | 4 (31) |
| Centrum semiovale | 5 (38) | 5 (38) |
| Corticospinal tracts | 5 (38) | 6 (46) |
| Periventricular | 5 (38) | 7 (54) |
| Subcortical | 2 (15) | 5 (38) |
| U-fibers | 1 (7.7) | 2 (15) |
| Dentate nucleus | 3 (23) | 0 |
| Brain region | Pre-HSCT, n (%) | Post-HSCT, n (%) |
|---|---|---|
| Asymptomatic (n = 5) | ||
| Brainstem | 2 (40) | 0 |
| Corpus callosum | 1 (20) | 0 |
| Corona radiata | 0 | 0 |
| Centrum semiovale | 1 (20) | 2 (40) |
| Corticospinal tracts | 0 | 0 |
| Periventricular | 0 | 4 (80) |
| Subcortical | 1 (20) | 1 (20) |
| U-fibers | 0 | 0 |
| Dentate nucleus | 1 (20) | 0 |
| Symptomatic (n = 13) | ||
| Brainstem | 5 (38) | 6 (46) |
| Corpus callosum | 6 (46) | 5 (38) |
| Corona radiata | 4 (31) | 4 (31) |
| Centrum semiovale | 5 (38) | 5 (38) |
| Corticospinal tracts | 5 (38) | 6 (46) |
| Periventricular | 5 (38) | 7 (54) |
| Subcortical | 2 (15) | 5 (38) |
| U-fibers | 1 (7.7) | 2 (15) |
| Dentate nucleus | 3 (23) | 0 |
Summary of neurophysiological outcomes from MRI, VEP, ABR, and NCV
| Test | Asymptomatic (%) | Symptomatic (%) | ||||
|---|---|---|---|---|---|---|
| Improved | Stable | Worsened | Improved | Stable | Worsened | |
| MRI | 40 | 60 | 0 | 0 | 54 | 46 |
| VEP | 25 | 50 | 25 | 33 | 33 | 33 |
| ABR | 80 | 0 | 20 | 17 | 67 | 17 |
| NCV | 60 | 20 | 20 | 10 | 80 | 10 |
| Test | Asymptomatic (%) | Symptomatic (%) | ||||
|---|---|---|---|---|---|---|
| Improved | Stable | Worsened | Improved | Stable | Worsened | |
| MRI | 40 | 60 | 0 | 0 | 54 | 46 |
| VEP | 25 | 50 | 25 | 33 | 33 | 33 |
| ABR | 80 | 0 | 20 | 17 | 67 | 17 |
| NCV | 60 | 20 | 20 | 10 | 80 | 10 |
Among the symptomatic children, half had stable results (n = 7; 54%) and the other half had signs of disease progression (n = 6; 46%; Table 6). Common regions with white matter abnormalities at the most recent examination were the periventricular (n = 7; 54%), brainstem (n = 6, 46%), and corticospinal tracts (n = 6; 46%; Table 5).
VEP
Thirteen patients had longitudinal VEP data. The majority of patients had stable (n = 5; 38%) or improved (n = 4; 31%) VEPs after transplantation. Among the 4 patients who had worsened VEPs, 2 had VEPs that increased in latency with no significant change in the amplitude of both eyes. At the most recent follow-up, 7 patients (54%) had normal VEPs and 6 (46%) had abnormal VEPs.
Among the asymptomatic children with data available, all but 1 improved (n = 1; 25%) or stabilized (n = 2; 50%; Table 6). The asymptomatic patient with worsened results was also the only asymptomatic child with an abnormal posttransplantation VEP.
Most symptomatic children improved (n = 3; 33%) or stabilized (n = 3; 33%; Table 6). At the most recent follow-up, 4 (57%) had normal VEPs and 3 (43%) had abnormal results.
Hearing assessment
Seventeen patients had longitudinal ABR data available. Most patients had improved (n = 6; 35%) or stabilized (n = 8; 47%) results. At the most recent follow-up, 10 patients (59%) had abnormal ABRs. The most common abnormality was prolonged latency (baseline: n = 12; 71%; follow-up: n = 11; 65%).
Except for 1 patient, all asymptomatic patients had improved ABR results (Table 6). Two patients had abnormal test results at the most recent follow-up. The asymptomatic child who had worsened ABR results also had an abnormal distortion product otoacoustic emission (DPOAE). This patient had normal hearing sensitivity in the right ear, but mild sensorineural hearing loss in the left ear.
For symptomatic patients, most improved (n = 2; 17%) or stabilized (n = 8; 67%) ABR results (Table 6). At the most recent examination, most had abnormal ABRs (n = 10; 83%).
None of the patients required hearing amplification but had delays in processing of auditory information. At baseline, all patients had a normal DPOAE, and at follow-up, only 1 had an abnormal DPOAE.
NCV
Fifteen patients had longitudinal NCV data. The majority of patients stabilized (n = 9; 60%) or improved (n = 4; 27%). At the most recent follow-up, 67% of patients had abnormal NCVs, along with 2 patients with minimally abnormal NCVs. Most patients had abnormalities in both motor and sensory nerves.
Except for 1 patient, all asymptomatic patients improved (n = 3; 60%) or stabilized (n = 1; 20%; Table 6). The patient with worsened results was noted to have a slight prolongation in the right peroneal F-wave and a slight reduction in peroneal conduction velocity. All other asymptomatic patients had normal NCV results at the most recent follow-up.
Among 10 symptomatic children, 1 patient had worsened NCVs and the remaining stabilized (n = 8; 80%) or improved (n = 1; 10%; Table 6). All but 1 patient had abnormalities present at the most recent examination. The patient whose NCV worsened died of disease progression 1 month after the NCV was measured.
EEG
Before transplantation, 3 patients (21%) had clinical seizures, and only 1 (7%) had epileptic changes in the EEG. After transplantation, 7 (47%) had clinical seizures, and 5 (33%) were verified as epileptic. All patients with seizures were symptomatic.
Discussion
Our study found that HSCT significantly alters the natural course of late-infantile Krabbe disease by improving lifespan and neurological outcomes. To our knowledge, this is the first study to exclusively and longitudinally assess neurodevelopment after transplantation of late-infantile patients with Krabbe disease, by using neurophysiologic, neuroradiologic, and functional outcomes.
After HSCT, patients experienced fluctuating psychosine concentrations. Nevertheless, there was a rapid decline in psychosine levels after HSCT. In contrast to this rapid decline, psychosine levels in dried blood spots in untreated early-infantile patients with Krabbe disease slowly declined after an initial increase.16
CSF levels also showed variability, with some patients experiencing a decline in levels and others an increase after HSCT. Although high CSF levels are important for diagnostic purposes, its significance in monitoring the disease status is not understood. In the natural history of this disease, there was also a wide range of values.1
Asymptomatic patients benefitted the most from transplantation. Longitudinal MRI, VEP, ABR, and NCV results demonstrated stabilization or improvement. This result is in stark contrast to that in untreated patients who showed progressive deterioration in neurophysiologic and neuroradiologic test results, demonstrating continually increasing latencies, diminished wave amplitude, slowed velocities, and increasing hyperintensities on brain MRI.1 The benefit of HSCT was also reflected in the patients’ functional outcomes; asymptomatic children had normal to near-normal development in all neurodevelopmental domains. Although there were few delays in the gross motor domain, all asymptomatic patients walked independently. Moreover, significant differences were found when comparing the developmental slopes of asymptomatic patients to those of the symptomatic and untreated patients.
As seen in neurophysiological tests, many symptomatic patients also had disease stabilization or improvement after HSCT. This outcome is a remarkable improvement compared with that in untreated patients who experienced severe demyelination and thus rapid worsening on MRI, NCV, VEPs, and ABR. Nevertheless, the functional outcomes of symptomatic patients depended on whether the patient had symptom onset after 12 months or before. Patients with onset at >12 months of age had delayed development but gained skills in all domains except in the motor area, whereas patients with onset at ≤12 months of age did not gain skills.
There were important differences in MRI, NCV, and ABR results between asymptomatic and symptomatic patients. ABR and NCV results in most asymptomatic patients improved after HSCT, whereas symptomatic patients stabilized. This suggests that early transplantation promotes disease improvement rather than stabilization. MRI results showed that all asymptomatic patients either improved or stabilized, whereas half of the symptomatic patients worsened, suggesting that benefits of early transplantation are most apparent in the brain. Both of these differences highlight the importance of performing transplantation in asymptomatic patients.
This study, along with the previous literature, saw limited and variable outcomes for gross motor function after transplantation.9,29 One explanation is that corticospinal tracts are the first structures to be myelinated and are the most heavily myelinated, making them particularly vulnerable to a GALC deficiency.30,31 Another explanation is that there may be differences in the degree of peripheral nerve involvement and thus, a difference in the response to treatment.
Five patients in this study underwent transplantation with the Pitt-RIC, the regimen specific for patients with metabolic disorders. Notably, all 5 patients survived and had full engraftment. Although the sample size was small, this regimen has been used in a prior study by Vander Lugt et al. In their report, 44 patients with nonmalignant disorders were treated with the Pitt-RIC regimen. Both the 5-year survival (85%) and engraftment rates were high.15 Combining the data from this study, prior studies that assessed similar RIC regimens, and our study, it appears that HSCT with RIC is safer, has good engraftment, and is well tolerated by patients with comorbidities.32,33
Our study supports the previous literature reclassifying late-infantile Krabbe disease as occurring at 12 to 36 months of age instead of the traditional 6 to 36 months. Bascou et al had found that the cohort with 6- to 12-month onset had a phenotype more similar to that of the early-infantile onset.1 Similarly, in our study, patients with onset from 6 to 12 months of age had worse developmental outcomes than those with onset at >12 months. This finding confirms that patients with disease onset at ≤12 months should be considered early infantile.
Because asymptomatic children benefitted the most from transplantation, this study highlights the importance of NBS, which allows for earlier diagnosis and increases the likelihood that children will undergo HSCT while asymptomatic. The challenge of implementing NBS for Krabbe disease has been in the difficulty of determining the disease onset and prognosis of the patient’s onset type. However, ongoing investigations evaluating psychosine as a biomarker for predicting the infantile phenotype has made NBS for Krabbe disease more feasible.16
Taking everything into consideration, we recommend that patients at high risk of developing infantile Krabbe disease, as determined by NBS and psychosine, be referred immediately to an experienced transplantation center for a comprehensive assessment with neurophysiological, neuroradiological, and neurodevelopmental measures. If it is confirmed that the patient will develop severe disease, the patient should be immediately worked up for transplantation while still asymptomatic. Because symptomatic patients with disease onset at ≤12 months of age have outcomes comparable to those of untreated patients, we recommend that these patients not be considered for transplantation. Symptomatic patients with onset at >12 months of age may still be eligible depending on the degree of neurodegeneration, although consideration should be given to those with onset between the ages of 9 and 12 months who are slow progressors, as reported by Bascou et al.1
Nevertheless, the choice to undergo transplantation is not trivial because there is still a risk of mortality. In addition, it is important to realize that from the time of the baseline neurodevelopmental evaluation until the engraftment of donor cells, there are a few weeks during which the disease continues to progress. However, without treatment, patients have a 100% risk of death, as the disease will progress rapidly. It is critical for physicians to consider all of these factors as well as the individual family’s circumstances when counseling.
A limitation of this study is the possibility of bias in patients recommended for transplantation. Because of the dangerous nature of transplantation, patients whose disease was too advanced were not recommended for the procedure. As a result, the benefits of HSCT are applicable to a less severe phenotype. In addition, because the study was conducted over 21 years, it is reasonable to presume that quality of care has improved, as knowledge of Krabbe disease and services for families have increased. Nevertheless, clinical management of patients was mostly consistent and followed guidelines set by our group in Escolar et al.34 Lastly, another study of 88 untreated patients with infantile Krabbe disease found no difference in survival when comparing patients diagnosed between 1999 and 2009 with those diagnosed between 2010 and 2018.35
In summary, we found that asymptomatic patients derive the most ```benefit from transplantation and have near normal neurodevelopment. However, unlike prior studies, we found that symptomatic patients also benefit from transplantation. Compared with their untreated counterparts, symptomatic transplant recipients demonstrated disease stabilization or improvement in neuroradiological tests. Furthermore, patients with symptom onset after 12 months of age continued to gain functional skills, whereas symptomatic patients with onset before 12 months of age did not.
Original data are available by e-mail request to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the families who traveled from other states to receive clinical services at the NDRD Program and chose to participate in this study; all the staff at the NDRD (University of Pittsburgh Medical Center); and the Duke Pediatric Blood and Marrow Transplant Program for making this study possible and the Legacy of Angels Foundation for continuing support of our work.
This research was partially funded by National Institutes of Health, National Institute of Neurological Disorders and Stroke grant R01NS061965-01 (M.L.E.), and the Legacy of Angels Foundation (M.L.E.).
Authorship
Contribution: I.C.Y. drafted the manuscript and contributed to the acquisition and analysis of the data; N.A.B. drafted a portion of the manuscript and contributed to the acquisition and analysis of the data; M.D.P. drafted a portion of the manuscript, conducted statistical analyses, and designed the figures; P.S. evaluated and observed the patients in follow-up, provided and reviewed transplantation data, and edited the manuscript; and M.L.E. provided oversight, conceived and designed the study, contributed to acquisition and analysis of the data, and edited the manuscript.
Conflict-of-interest-disclosure: M.L.E. and P.S. have financial interest in Forge Biologics, and M.L.E. is a part-time employee. The remaining authors declare no competing financial interests.
Correspondence: Maria L. Escolar, Department of Pediatrics, University of Pittsburgh, 4401 Penn Ave, Pittsburgh, PA 15144; e-mail: maria.escolar@chp.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal