


Abstract
Plasma cells no longer express a B-cell antigen receptor and are hence deprived of signals crucial for survival throughout B-cell development. Instead, normal plasma cells, as well as their malignant myeloma counterparts, heavily rely on communication with the bone marrow (BM) microenvironment for survival. The plasma cell heparan sulfate proteoglycan (HSPG) syndecan-1 (CD138) and HSPGs in the BM microenvironment act as master regulators of this communication by co-opting specific growth and survival factors from the BM niche. This designates syndecan-1/HSPGs and their synthesis machinery as potential treatment targets in multiple myeloma.
Introduction
From the pre–B-cell stage onward, all steps of B-cell development critically depend on membrane expression of the B-cell antigen receptor (BCR), which provides essential signals for B-cell survival, proliferation, and differentiation.1,2 Upon plasma cell differentiation, however, the BCR is no longer membrane expressed but secreted as immunoglobulin. The consequent loss of BCR-derived survival signals necessitates a complete makeover of the cellular signaling-machinery to enable plasma cells to survive. As discussed in this review, the transmembrane heparan sulfate proteoglycan (HSPG) syndecan-1/CD138 is an essential component of this B-cell to plasma cell metamorphosis. In collaboration with HSPGs expressed by bone marrow (BM) stromal cells, syndecan-1 allows normal and myeloma plasma cells to efficiently co-opt soluble factors produced by cells in the BM niche. This promotes plasma cell homing/retention and survival, as well as multiple myeloma (MM) tumor growth. Strategies to target HSPG/syndecan-1 in MM are discussed.
HSPGs
HSPGs are proteins with ≥1 covalently attached HS glycosaminoglycan (GAG) chains, which in the native form consist of alternating N-acetylglucosamine (GlcNAc) and D-glucuronic acid (GlcA) units.3,4 These macro-molecules are expressed in all mammalian tissues as extracellular matrix components (eg, perlecan) or as cell membrane–bound glycoproteins (eg, syndecans and glypicans). To execute their function, the HS chains undergo a complex series of modifications, comprising N-deacetylation/N-sulfation, epimerization, and O-sulfation at different positions (Figure 1A). This endows HS chains with highly modified domains, which provide specific docking sites for a large number of bioactive molecules. Binding of these ligands, such as growth factors, cytokines, and chemokines, serves a variety of functions, ranging from immobilization and concentration to distinct modulation of signaling, facilitating the formation of receptor complexes (Figure 1B).5-11 By regulating the activity of these protein ligands, HSPGs mediate important biological processes, including cell growth, adhesion, migration, angiogenesis, and tissue remodeling, which are activities crucial in, among others, the control of embryonic development and immunity and the pathogenesis of cancer.4,10,12-14
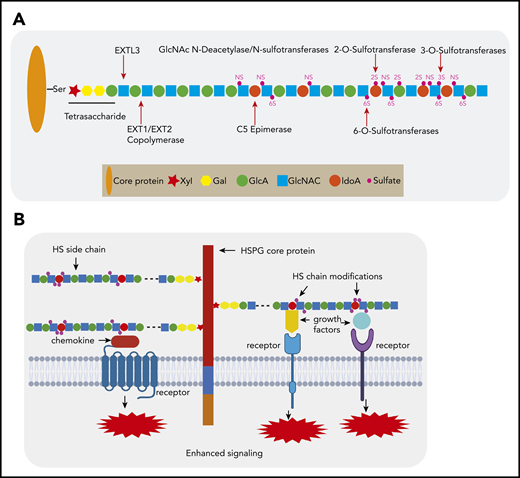
HSPG structure and HS chain modification. (A) HSPGs consist of a core protein to which 1 or more HS side chains are covalently attached. The synthesis of a HS chain starts with the attachment of a tetrasaccharide linker (xylose–galactose–galactose–glucuronic acid) to a serine residue of the core protein. The enzyme EXTL3 adds the first GlcNAc to the tetrasaccharide linker. The co-polymerases EXT1/EXT2 mediate the elongation of the HS chain by adding GlcA-GlcNAc residues. Subsequently, the HS chains undergo various modifications, which include GlcNAc deacetylation and sulfation by N-deacetylase/N-sulfotransferases, epimerization of GlcA to its epimer iduronic acid by glucuronyl C5-epimerase, and sulfation at the 2-O-position, 3-O-position, and C-6-position by different sulfotransferases. After completion of this synthesis and modification process, the structure and function of the HS chain can still be further modified by a number of endo- and exo-enzymes. These include heparanase, which is expressed in damaged tissues and cancer but hardly by normal tissues and can cleave HS chains into fragments. Furthermore, the sulfation status of HS, which is crucial for protein interactions, can be modified by sulfatase-1 (Sulf-1) and sulfatase-2 (Sulf-2), which remove the 6-O sulfate moieties from disulfated GlcA-GlcNS6S and trisulfated IdoA2S-GlcNS6S. (B) HSPGs bind multiple growth factors, cytokines, and chemokines via their HS side chains and facilitate the interaction of these ligands with their cognate receptors, thereby regulating the activities of these proteins and leading to enhanced signaling activation. Importantly, HS modifications determine the binding capacity and specificity of HS chains for a given protein and are both cell type and differentiation stage specific.
HSPG structure and HS chain modification. (A) HSPGs consist of a core protein to which 1 or more HS side chains are covalently attached. The synthesis of a HS chain starts with the attachment of a tetrasaccharide linker (xylose–galactose–galactose–glucuronic acid) to a serine residue of the core protein. The enzyme EXTL3 adds the first GlcNAc to the tetrasaccharide linker. The co-polymerases EXT1/EXT2 mediate the elongation of the HS chain by adding GlcA-GlcNAc residues. Subsequently, the HS chains undergo various modifications, which include GlcNAc deacetylation and sulfation by N-deacetylase/N-sulfotransferases, epimerization of GlcA to its epimer iduronic acid by glucuronyl C5-epimerase, and sulfation at the 2-O-position, 3-O-position, and C-6-position by different sulfotransferases. After completion of this synthesis and modification process, the structure and function of the HS chain can still be further modified by a number of endo- and exo-enzymes. These include heparanase, which is expressed in damaged tissues and cancer but hardly by normal tissues and can cleave HS chains into fragments. Furthermore, the sulfation status of HS, which is crucial for protein interactions, can be modified by sulfatase-1 (Sulf-1) and sulfatase-2 (Sulf-2), which remove the 6-O sulfate moieties from disulfated GlcA-GlcNS6S and trisulfated IdoA2S-GlcNS6S. (B) HSPGs bind multiple growth factors, cytokines, and chemokines via their HS side chains and facilitate the interaction of these ligands with their cognate receptors, thereby regulating the activities of these proteins and leading to enhanced signaling activation. Importantly, HS modifications determine the binding capacity and specificity of HS chains for a given protein and are both cell type and differentiation stage specific.
Syndecan-1 in the metamorphosis of B cells to plasma cells
During terminal differentiation, B-lineage cells acquire syndecan-1 as an integral part of the plasmacytic gene expression program, driven by the transcription factors Blimp-1 and XBP1.15 Syndecan-1 is a transmembrane HSPG and is used in clinical practice as canonical marker for normal and myeloma plasma cells. Functional studies have unequivocally demonstrated that syndecan-1 is crucial for effective communication of plasma cells with their BM niche, acting as a versatile coreceptor for chemokines, cytokines, and growth factors, mediating plasma cell homing, survival, and, in MM, tumor growth.13,16-18 A landmark study by McCarron et al18 demonstrated that mice with a B lineage–intrinsic syndecan-1 deletion show a defective long-term humoral immunity. This defect could be traced to the inability of syndecan-1–negative plasma cells to respond to interleukin-6 (IL-6)- and a proliferation-induced ligand (APRIL)–mediated survival signals. Attenuated APRIL signaling has similarly been implicated in the humoral immune defect of mice deficient in Glce, encoding the HS-modifying enzyme glucoronyl-C5-epimerase.19 This enzyme converts D-glucuronic acid (GlcA) into its stereo-isomer L-iduronic acid (IdoA), thereby enhancing the flexibility of the HS chain, which provides access for ligands to specific binding regions.19,20 Mice with a B lineage–intrinsic Glce deletion displayed diminished antibody levels and plasma cell numbers, whereas Glce-deficient plasma cells were unable to bind APRIL and failed to respond to APRIL-mediated survival signals.19
These studies show that the activity of IL-6 and APRIL, credibly the 2 foremost prosurvival factors for long-lived plasma cells, is controlled by syndecan-1 and requires specific HS chain modifications. For IL-6, the exact mode of interaction with syndecan-1 has not yet been defined, but for tumor necrosis family growth factor APRIL, the mechanism is firmly established. APRIL is secreted in the BM microenvironment by various cell types, including osteoclasts,21 eosinophils,22 and megakaryocytes,23,24 and binds the HS moieties of syndecan-1 via its N-terminal lysine-rich region. This leaves the tumor necrosis factor–like region of APRIL free to interact with the receptors' B-cell maturation antigen and transmembrane activator and calcium-modulator and cyclophilin ligand interactor.8,25 Interestingly, unlike its close relative B-cell activating factor, which automultimerizes, APRIL strictly requires interaction with HS moieties to form multimers at the plasma cell membrane and generate signals that mediate plasma cell longevity.26,27 This HS dependency defines APRIL as a plasma cell survival factor specifically designed to collaborate with syndecan-1.
Syndecan-1 in the pathogenesis of MM
In addition to its physiologic role in plasma cells, syndecan-1 also plays a key role in the pathogenesis of MM, promoting tumor cell survival and proliferation. By using a SCID-hu mouse xenotransplant model, Yang et al28 demonstrated that knockdown of syndecan-1 strongly attenuates MM growth. Treatment with bacterial heparinase III, an enzyme that degrades HS, also inhibited tumor growth, suggesting involvement of the HS moieties of syndecan-1. The key role of these HS chains was corroborated by Reijmers et al,17 using short hairpin RNAs targeting EXT1, an enzyme crucial for HS chain synthesis. EXT1 knockdown, and consequent loss of cell surface HS, completely prevented the outgrowth of MM xenotransplants, whereas EXT1 knockdown in already established MM xenotransplants resulted in massive tumor necrosis.17 Furthermore, in a murine MM model, treatment with an anti–syndecan-1 antibody enhanced sensitivity to bortezomib chemotherapy.29 Together, these studies show the critical importance of syndecan-1 for MM cell survival and growth and provide proof of concept for the therapeutic potential of strategies targeting syndecan-1 or its HS chains.
In line with its role in mediating plasma cell survival, syndecan-1 promotes, or is even essential, for the activity of many MM survival/growth factors. These include the already discussed major plasma cell survival factors IL-6 and APRIL,25,30 but, in addition, comprise factors that are aberrantly expressed by MM cells or by the microenvironment, acting as auto- and/or paracrine growth factors. These mediators include hepatocyte growth factor (HGF),7,31 insulin-like growth factor-1,32 and several epidermal growth factor (EGF) family proteins with HS-binding properties, including heparin-binding EGF, neuregulin-1, and amphiregulin.9,33 Interaction of these factors with the HS moieties of syndecan-1 enhances RAS/MAPK and phosphatidylinositol 3-kinase/protein kinase B pathway activation, thereby promoting MM cell growth and survival.7,9,31-33
A distinct oncogenic signaling pathway that also involves syndecan-1 is Wnt/β-catenin signaling.34 Aberrant Wnt signaling, activated by auto- and paracrine Wnt ligands, is common in MM and promotes tumor growth, acting upstream of c-MYC and cyclin D1.35-39 We recently demonstrated that syndecan-1 binds Wnts and R-spondins, produced in the BM microenvironment by MM cells, bone marrow stromal cells (BMSCs), and osteoblasts. This binding, which involves the HS moieties of syndecan-1, facilitates the interaction of these ligands with their respective receptors (ie, with Frizzleds and LGR4),11 thereby enhancing Wnt signaling and proliferation.
Notably, the tumorigenic role of syndecan-1 in MM is not restricted to its function as a membrane-bound signaling mediator: MM cells show strongly enhanced syndecan-1 shedding, promoted by heparanase, resulting in accumulation of syndecan-1 and of HS cleavage products in the BM microenvironment, stimulating migration, and acting as reservoir for survival/growth factors.28,40-42 Importantly, these factors do not only comprise mediators that directly interact with MM cells but also factors like fibroblast growth factor-2 and vascular endothelial growth factor, which promote angiogenesis and, hence, contribute to a tumor-permissive microenvironment.43-45
Apart from mediating tumor growth and survival, syndecan-1 has also been implicated in osteolytic bone disease, a major MM disease complication causing severe morbidity. Osteolytic lesions in MM are initiated by a disturbed balance between osteoblast-mediated bone formation and bone resorption by osteoclasts, a balance that is critically dependent on the receptor activator of nuclear factor-κB ligand (RANKL)/RANK pathway, which controls osteoclast activity.46 Interestingly, syndecan-1 on the MM cell surface was shown to bind, internalize, and degrade the RANKL antagonist osteoprotegerin, thereby promoting RANKL-mediated osteoclastogenesis, osteoclast activity, and osteolysis.47 Furthermore, RANKL/RANK-mediated osteoclast activity may also be promoted by soluble syndecan-1: Shed syndecan-1 was reported to bind HGF and to promote HGF/MET signaling in osteoblasts, leading to enhanced RANKL expression and osteoclastogenesis.48,49
HSPGs expressed by cells in the plasma cell and MM BM niche
HSPGs are not only expressed by normal and MM plasma cells but also by BMSCs, endothelial cells, and osteoblasts,50-52 and, interestingly, have been implicated in controlling the homing of hematopoietic progenitor cells to, and retention, in the BM.50,53 Within this microenvironment, long-lived plasma cells are located in close contact with BMSCs expressing high levels of the chemokine CXCL12, also referred to as CXCL12 abundant reticular (CAR) cells.54 These niche cells strongly express HSPGs55,56 ; however, the role of these HSPGs in the interaction of normal and MM plasma cells with the BM niche has remained unexplored. We recently found that human CAR-like (HS5) cells constitutively carry HS-bound CXCL12 on their cell surface. CRISPR/Cas9-mediated deletion of the HS polymerase EXT1 led to loss of HS and a concurrent loss of membrane-bound CXCL12. This strongly attenuated the ability of the CAR cells to support adhesion of MM cells and protect these cells from bortezomib-induced death. Interestingly, the γ-isoform of CXCL12 was found to control these interactions.57 Unlike the commonly studied CXCL12 isoform CXCL12α, CXCL12γ possesses an extended C-terminal domain, which contains 3 positively charged BBXB motives that convey an extraordinarily high affinity for HS,58,59 explaining its membrane retention on secretion. These findings suggest an intriguing scenario in which differences in affinity for HS entail various CXCL12 isoforms with distinct roles in the regulation of plasma and MM cell homing and retention in the BM: unlike CXCL12α, which creates a chemo-attractive gradient guiding plasmablasts and MM cells to their BM niches, the membrane-bound CXCL12γ isoform may function as a niche chemokine, controlling plasma and MM cell adhesion/retention, survival, and proliferation. Further studies are needed to explore this scenario.
Syndecan-1/HSPGs and their synthesis machinery as targets for the treatment of MM
The important role of HSPGs in the biology of MM and other human cancers designates HSPGs and their synthesis and modification enzymes as potential therapeutic targets. Possible targeting strategies include monoclonal antibodies directed against HSPG core proteins or HS side chains, various heparin mimetics to block HSPG–ligand interaction, and small molecule inhibitors targeting enzymes involved in HS synthesis or modification. Although preclinical studies have provided robust prove of concept for the feasibility and therapeutic potential of these strategies, clinical translation is still largely limited to early phase studies.
The antibody–drug conjugate indatuximab ravtansine (BT062), targeting the syndecan-1 core protein, was shown to inhibit tumor growth in a MM xenograft model, leading to prolonged host survival.60 In this setting, BT062 acts additively, or even synergistically, with clinically approved therapies for MM.61 In a first-in-human clinical phase 1/2a study, in 32 patients with relapsed and/or refractory MM, more than 75% of heavily pretreated patients achieved stable disease or better, with minor and partial responses occurring in 14.7% of patients, supporting further investigation as part of a combination regimen.62 Indeed, a phase 1/2a clinical study in relapsed and refractory MM with BT062, in combination with lenalidomide or pomalidomide and dexamethasone, has been completed, but the results have not yet been reported (NCT01638936). In addition, CAR T cells incorporating the single chain variable fragment (scFv) of BT062 have been generated and have shown safety and therapeutic efficacy in a mouse myeloma xenotransplant model.63 Based on these findings, a phase 1 clinical study with these CD138 CAR T cells has been initiated in patients with refractory and relapsed MM (NCT03672319).
Heparin, a highly sulfated form of HS, has been previously shown to have potent antitumor activity in several experimental and clinical studies, but its strong anticoagulant activity has impeded its use as an antitumor drug.44,64 This has incited the development of several heparin mimetics with reduced anticoagulant activity. These compounds function by inhibiting heparanase and by competing with HS for binding of essential growth and survival factors.64,65 The heparin mimetic roneparstat (SST0001) has been shown to inhibit myeloma growth in xenograft models by disrupting of the heparanase–syndecan-1 axis.66,67 Additional preclinical studies demonstrated that roneparstat reduced MM-induced osteoclast differentiation and bone destruction and repressed myeloma cell stemness and drug resistance.68,69 A phase 1 clinical study with roneparstat in patients with advanced MM has recently been completed (NCT01764880). The results show a favorable safety profile but very limited antitumor activity as single agent.70
In addition to the HSPG itself, the enzymes of the HS synthesis and modification machinery present potentially promising therapeutic targets. HSPG sulfation in particular, is a major determinant of the affinity and specificity of the interaction of protein ligands with HS.3,10,71 Interestingly, the small molecule inhibitor OKN-007, which targets Sulf-2, an enzyme regulating HS sulfation, has antitumor activity in several different types of cancer.72-75 Recently, a phase 2 clinical study with this compound has been initiated in patients with recurrent glioblastoma (NCT04388475).
Conclusions and perspectives
Syndecan-1, in concert with HSPGs expressed by BMSCs, plays a pivotal role in the interaction of normal and MM plasma cells with the BM niche, by co-opting chemokines, cytokines, and growth/survival factors, which endorse the homing, adhesion/retention, and survival of normal plasma cells, and mediate MM cell growth and drug resistance (Figure 2).

Syndecan-1 and BM stromal HSPGs in the interaction of MM plasma cells with the BM microenvironment. Schematic representation of a MM plasma cells interacting with different components of the BM microenvironment. Specialized, CAR-like niche cells in the BM microenvironment secrete high levels of CXCL12α, which steers (MM) plasma cells migration/homing to the BM. The distinct cell types in the BM microenvironment, including BMSCs, hematopoietic cells, osteoblasts, and osteoclasts, as well as the MM cells themselves, produce various survival/growth factors and cytokines. Membrane-bound syndecan-1 can bind many of these soluble factors and present them to their cognate receptors, thereby promoting MM cell proliferation and survival. Furthermore, syndecan-1 shed by MM cells may act as a reservoir for soluble factors, including mediators of angiogenesis and osteoclastogenesis. In addition, by expressing HSPG-bound CXCL12γ on their cell membrane, CAR-like stromal niche cells can engage the chemokine receptor CXCR4 on MM cells, promoting MM cell retention, survival, and drug resistance.
Syndecan-1 and BM stromal HSPGs in the interaction of MM plasma cells with the BM microenvironment. Schematic representation of a MM plasma cells interacting with different components of the BM microenvironment. Specialized, CAR-like niche cells in the BM microenvironment secrete high levels of CXCL12α, which steers (MM) plasma cells migration/homing to the BM. The distinct cell types in the BM microenvironment, including BMSCs, hematopoietic cells, osteoblasts, and osteoclasts, as well as the MM cells themselves, produce various survival/growth factors and cytokines. Membrane-bound syndecan-1 can bind many of these soluble factors and present them to their cognate receptors, thereby promoting MM cell proliferation and survival. Furthermore, syndecan-1 shed by MM cells may act as a reservoir for soluble factors, including mediators of angiogenesis and osteoclastogenesis. In addition, by expressing HSPG-bound CXCL12γ on their cell membrane, CAR-like stromal niche cells can engage the chemokine receptor CXCR4 on MM cells, promoting MM cell retention, survival, and drug resistance.
Preclinical studies have provided robust prove of concept for therapeutic efficacy of targeting syndecan-1/HSPGs, as well as some of the enzymes involved in HS chain modification, in various types of cancer, including MM. The thus far limited data from early phase clinical studies suggest that targeting of HSPGs is well tolerated, but further studies are needed to allow a verdict regarding therapeutic efficacy.
For the design of future therapeutics, the fact that distinct HS chain modifications create binding sites for specific ligands is of particular relevance. Characterization of the HS modifications that facilitate binding of crucial MM growth and survival factors, such as APRIL, IL-6, and WNTs, will not only define these HS motives but also identify the enzymes responsible for their synthesis. This knowledge can be exploited to design (allosteric) inhibitors interfering with the HS–ligand interaction, as well as small molecule inhibitors targeting specific HS-modifying enzymes, a strategy that may finetune and improve the clinical safety and efficacy of syndecan-1/HSPG targeted therapies.
Acknowledgments
This work was supported by grants from the Dutch Cancer Society and Lymph&Co (S.T.P. and M.S.) and a scholarship from the China Scholarship Council (Z.R.).
Authorship
Contribution: Z.R., M.S., and S.T.P. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Steven T. Pals, Department of Pathology, Amsterdam University Medical Centers, Loc. AMC, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: s.t.pals@amsterdamumc.nl; and Zemin Ren, Department of Pathology, Amsterdam University Medical Centers, Loc. AMC, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: renzemin12@hotmail.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal