

In this issue of Blood, provide a detailed phenotypic and functional analysis of the inflammatory infiltrate in the Langerhans cell histiocytosis (LCH) lesion. Using a BRAFV600ECD11C preclinical model of systemic LCH, they demonstrate the synergistic effect of dual treatment with a MAPK inhibitor directed toward the pathologic LCH cell and checkpoint blockade with an anti–programmed death-1 (α-PD-1) antibody directed toward the exhausted T cells in the LCH lesion microenvironment (see figure).1
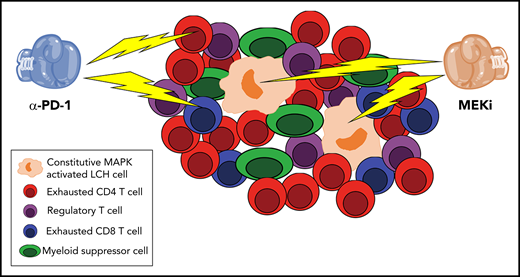
Double trouble for LCH lesions. Sengal et al perform a detailed phenotypic and functional analysis of the inflammatory infiltrate in LCH lesions. Confirming and extending prior observations, Sengal et al show that the constitutively active MAPK LCH cells are a minority of the population, whereas exhausted CD4 and CD8 T cells, regulatory T cells, and myeloid-derived suppressor cells predominate. Using a BRAFV600ECD11C preclinical model of systemic LCH, they demonstrate that α-PD-1 and MEKi have synergistic activity in reducing the size of the LCH lesions.
Double trouble for LCH lesions. Sengal et al perform a detailed phenotypic and functional analysis of the inflammatory infiltrate in LCH lesions. Confirming and extending prior observations, Sengal et al show that the constitutively active MAPK LCH cells are a minority of the population, whereas exhausted CD4 and CD8 T cells, regulatory T cells, and myeloid-derived suppressor cells predominate. Using a BRAFV600ECD11C preclinical model of systemic LCH, they demonstrate that α-PD-1 and MEKi have synergistic activity in reducing the size of the LCH lesions.
LCH is caused by clonal expansion of myeloid precursors that differentiate into CD1a+/CD207+ cells.2,3 Identification of BRAFV600E mutations or other mutations that result in constitutive activation of the MAPK pathway has led to characterization of LCH as an inflammatory myeloid neoplasm.4,5 LCH can present at all ages and with a spectrum of findings from single-system involvement to widespread, multisystem disease. Therapy is risk-stratified based on extent of disease.2,3 Those with multisystem disease or high-risk single system involvement require systemic chemotherapy. Despite progress in the treatment of patients with LCH, disease reactivation rates remain >30%, and mortality rates for patients with organ dysfunction may reach 20%.2,3 Long-term sequalae of disease are also significant.2,3 The identification of universal MAPK pathway activation in LCH paved the way for treatment with targeted inhibitors.6-8 Although response to BRAF or MEK inhibitors is generally excellent, recurrence of disease upon discontinuation of therapy is almost universal.8,9
This is where the work of Sengal et al comes in. Similar to Reed-Sternberg cells in Hodgkin disease, prior work has demonstrated that the pathologic LCH cell constitutes a minority of the cellular population within the lesion.2,3 Prior studies have characterized the surrounding inflammatory infiltrate and demonstrated a preponderance of M2-polarized macrophages, T cells, and regulatory T cells as well as increased expression of program death ligand 1 (PD-L1) on the LCH cells.4,8-10 Sengal et al take this a step farther and use cytometry by time-of-flight (CYTOF) coupled with functional assays to more thoroughly characterize the inflammatory infiltrate in LCH. They confirm that the LCH lesional CD207+ cells express high levels of PD-L1 and that the inflammatory infiltrate comprises dendritic cells, myeloid-derived suppressor cells, and T cells. Careful analysis of the T cells’ compartment revealed skewing toward CD4+ T cells and regulatory T cells relative to CD8+ T cells. Both CD4+ and CD8+ T cells exhibited signs of T-cell exhaustion with increased expression of inhibitor receptors, including PD-1, TIM-3, and LAG-3, decreased cytokine production upon stimulation, and decreased effector function. Importantly, the exhausted phenotype could be reversed with PD-1 blockade. The mechanistic basis for the preponderance of exhausted T cells remains unclear, although chronic antigen stimulation by the LCH lesional cells themselves and/or immunosuppressive microenvironment mediated by infiltrating myeloid-derived suppressor cells, regulatory T cells, or the LCH cells themselves are possibilities.
To begin to evaluate whether checkpoint blockade might have clinical utility, the authors move to a BRAFV600ECD11C preclinical murine model of LCH. After documenting that it recapitulates the inflammatory infiltrate and exhausted T-cell phenotype seen in human LCH lesions, they randomize cohorts of mice to treatment with the MEK-1 inhibitor trametinib or the checkpoint inhibitors PDL-1 or PD-1. Although PD-L1 blockade had minimal effects, monotherapy with α-PD-1 or trametinib resulted in partial responses. Interestingly, trametinib led to loss of the LCH lesional cells but a concomitant increase in infiltrating CD3+ T cells. In contrast, α-PD-1 resulted in a significant decrease in infiltration lymphoid cells with nominal effect on myeloid cells. Strikingly, combined therapy with trametinib and α-PD-1 resulted in a synergistic effect on disease burden. A shortcoming of the study was the lack of survival analyses evaluating the global impact of the therapy. Studies looking at optimal length of therapy to prevent recurrence or whether combination with traditional chemotherapy is beneficial are also needed. Nonetheless, this work highlights the potential value of targeting not only the pathologic LCH CD207A cell but also the abnormal microenvironment in which it resides. Clinical trials evaluating dual checkpoint blockade and MAPK inhibition are warranted.
Conflict-of-interest disclosure: The author has served on advisory boards from Sobi and Novartis and currently serves on the data safety monitory committee for NovaImmune.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal