

Key Points
GC B-cell expression of NrasQ61R and MYC drives a highly malignant MM.
The VQ MM model serves as a robust platform for preclinical studies of novel therapeutic agents.

Abstract
NRASQ61 mutations are prevalent in advanced/relapsed multiple myeloma (MM) and correlate with poor patient outcomes. Thus, we generated a novel MM model by conditionally activating expression of endogenous NrasQ61R and an MYC transgene in germinal center (GC) B cells (VQ mice). VQ mice developed a highly malignant MM characterized by a high proliferation index, hyperactivation of extracellular signal-regulated kinase and AKT signaling, impaired hematopoiesis, widespread extramedullary disease, bone lesions, kidney abnormalities, preserved programmed cell death protein 1 and T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domain immune-checkpoint pathways, and expression of human high-risk MM gene signatures. VQ MM mice recapitulate most of the biological and clinical features of human advanced/high-risk MM. These MM phenotypes are serially transplantable in syngeneic recipients. Two MM cell lines were also derived to facilitate future genetic manipulations. Combination therapies based on MEK inhibition significantly prolonged the survival of VQ mice with advanced-stage MM. Our study provides a strong rationale to develop MEK inhibition–based therapies for treating advanced/relapsed MM.
Introduction
Multiple myeloma (MM), a tumor of mature plasma B cells that produces antibodies,1 ranks as the second most common blood malignancy in the United States with an estimated 32 270 new cases in 2020 (https://www.cancer.org). Myeloma incidence has been steadily growing in both male and female cases since the mid-70’s worldwide (Surveillance, Epidemiology, and End Results [SEER] statistics and Cowan et al2 ). With the introduction of “novel agents” to MM treatment, including proteasome inhibitors, immunomodulatory agents, and therapeutic antibodies, MM patient survival has increased (5-year survival now exceeds 50%), and quality of life has also improved.3 Relapses are almost inevitable and clinical benefit becomes less durable with each successive regimen until ultimate demise from multidrug-resistant MM4 and/or aggressive manifestations such as nonsecretory MM, extramedullary disease, and plasma cell leukemia.5
Aberrant MYC expression is an early molecular event that associates with progression from indolent monoclonal gammopathy of undetermined significance (MGUS) to overt MM.6,7 Consistent with this observation in humans, activation of a human MYC transgene in germinal center (GC) B cells induces a highly penetrant, indolent MM in Vκ*MYC mice.8 Extramedullary involvement occurs in ∼30% of Vκ*MYC mice. Targeting MYC (eg, through bromodomain inhibition) has been shown to confer significant benefit.9
The infrequency of advanced MM in the Vκ*MYC model suggests that additional genetic mutations are required. Indeed, genomic analysis of Vκ*MYC myeloma tumors identified frequent aneuploidy and biallelic deletions of Rb1, Kdm6a, or Cdkn2a.10 Large-scale sequencing analyses reveal key progression events that primarily drive the hyperactivation of the RAS/RAF/MEK/extracellular signal-regulated kinase (ERK) pathway and, secondarily, the NF-κB pathway.11-14 RAS pathway mutations collectively constitute >50% of advanced and relapsed MM cases14 and are even more frequent in drug-resistant patients (72%15 ). Here, we present the first genetically engineered Ras; MYC-driven MM murine model (VQ mice). VQ mice recapitulate most of the biological and clinical features of human advanced/high-risk MM.
Methods
All mouse lines were maintained in a pure C57BL/6 genetic background (>N10). The Vκ*MYC, conditional oncogenic NrasLox-stop-Lox (LSL) Q61R/+ (Nras), and immunoglobulin G1 (IgG1)-Cre mice were described previously.8,16-18 CD45.1+ transplant recipients were purchased from The Jackson Laboratory and bred at the Biotron Animal Research Services facility at the University of Wisconsin (UW)-Madison. All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD) and approved by the Animal Care and Use Committee at UW-Madison. The program is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Additional methods are described in supplemental Methods (available on the Blood Web site).
Results
Generation of an oncogenic Nras; MYC-driven MM model
Mutations in RAS pathway genes (NRAS, KRAS, and BRAF) are rare in MGUS but are collectively found in >50% of patients with advanced or relapsed MM.11,13,14,19 We and others characterized multiple oncogenic Ras models, including KrasG12D/+,20-22 NrasG12D/+,23,24 NrasG12D/G12D,25,26 and NrasQ61R/+.27 When activated throughout the hematopoietic system using Mx1-Cre, we only observed B-cell malignancies in a fraction of oncogenic Nras mice but never in oncogenic Kras mice.22,25,28 Moreover, unlike the prevalence of NRASG12 and G13 mutations in myeloid diseases, NRAS Q61 mutations predominantly occur in MM27 and have been reported to associate with MM poor prognosis.29 Therefore, we chose the NrasQ61R/+ allele16 as the MM progression driver.
Chesi et al reported that Vκ*MYC mice develop a highly penetrant, indolent MM,8 consistent with the role of dysregulated MYC expression in MM initiation.6 The Vκ*MYC allele includes multiple copies of the Vκ*MYC transgene arranged in head-to-tail order8,10 (supplemental Figure 1). Each copy contains an engineered stop codon at the third amino acid and a floxed 3′ κ enhancer. The stop codons could be sporadically reverted by activation-induced cytidine deaminase in GC B cells. The floxed 3′ κ enhancer could be removed in the presence of Cre recombinase to downregulate MYC expression. Given that MYC activation signatures are overexpressed upon transition from MGUS to overt myeloma30 and NRAS Q61 mutations are prevalent in the latter, we reasoned that the Vκ*MYC allele, when combined with the NrasQ61R/+ allele, may allow in vivo selection of myeloma-initiating cells with an appropriate level of MYC expression. The level of MYC expression may be critical as previous attempts utilizing an unfloxed Vκ*MYC allele in combination with various oncogenes almost invariably generated aggressive B-cell lymphoma (M.C., unpublished data), except for the extramedullary MM noted in Vκ*MYC mice crossed with the Eμ-BCL2 allele.8
We used IgG1-Cre18 to drive NrasQ61R/+ expression and randomly select for hMYC expression levels in GC B cells (Figure 1A). In addition to the Vκ*MYC; NrasLSL Q61R/+; IgG1-Cre (VQ) compound mice, we also generated NrasLSL Q61R/+; IgG1-Cre (Q), Vκ*MYC; IgG1-Cre (V) mice. IgG1-Cre mice served as control throughout the study. To boost IgG1-Cre expression, 4-hydroxy-3-nitrophenylacetyl–chicken γ globulin (NP-CGG) was used to immunize 6- to 7-week-old mice. We closely monitored the control, Q, V, and VQ mice via regular serum protein electrophoresis (SPEP). A significant fraction of Q, V, and VQ mice developed an M spike over time (Figure 1B) and died prematurely (Figure 1C). In the VQ cohort, 14 of 17 VQ mice developed an M spike. Ultimately, 4 VQ mice were found dead and could not be evaluated. In the remaining 13 VQ mice, 8 of 13 died of a highly malignant MM, characterized by increased CD138+B220− myeloma cells (>10%) in bone marrow (BM), spleen, and/or lymph node (LN) and infiltration of myeloma cells to liver and kidney (Figure 1D; supplemental Figure 2A; Figure 1E; supplemental Figure 6A). Immunoglobulin deposition in kidney (Figure 1E) and rouleaux formation in peripheral blood (supplemental Figure 2B) were also seen in some of the VQ MM mice. Associated with MM-like phenotypes, VQ mice showed impaired hematopoiesis typically seen in advanced MM patients, including anemia and thrombocytopenia (Figure 1F). Isotyping of serum antibodies in 5 VQ MM mice demonstrated detectable κ light chain in all mice along with IgG2b (B2518-VQ1), IgA and IgG3 (B2519-VQ2, B3518-VQ5, and B3520-VQ4), or λ light chain (B2576-VQ3) (supplemental Table 1). Consistent with the isotyping results, our clonality analysis using a D-J rearrangement assay31 showed that VQ1 myeloma had a single dominant clone whereas VQ2 myeloma was oligoclonal (Figure 1G). For the remaining 5 of 13 VQ mice, they either developed other lymphoid diseases (n = 3) (eg, B-cell lymphoma in B2509) or died without a defined hematologic malignancy (n = 2) (eg, B3300) (Figure 1C). Because both B2509 and B3300 showed an M spike in SPEP analysis, we isotyped their serum samples; both were positive for κ light chain, IgM, and a few other IgGs (supplemental Table 1).
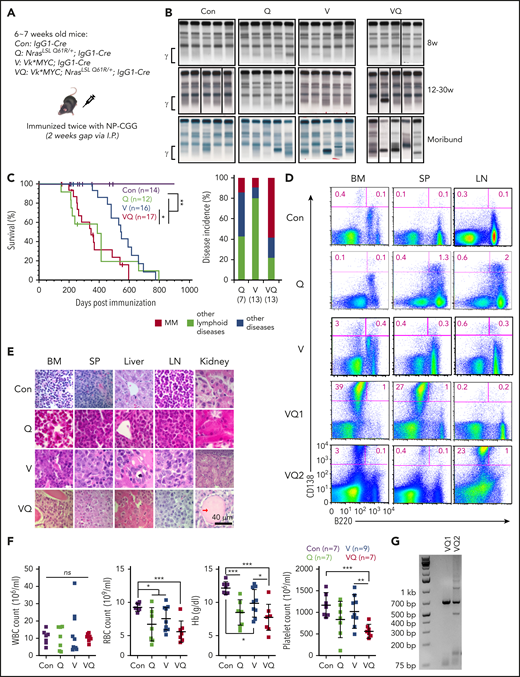
A fraction of NrasQ61R/+; Vκ*MYC mice develop MM. (A) Acronyms of transgenic mouse lines and illustration of IgG1-Cre induction scheme. (B) SPEP was performed on representative mice from each group serially bled at the indicated weeks. The brackets show the γ-globulin component of the serum. (C) Kaplan-Meier survival curves (left) and disease incidences (right) of different groups of animals. MM is defined as CD138+B220− cells being >10% in BM, spleen, and/or LN along with end-organ abnormalities. (D) Flow cytometric analysis of B220 and CD138 expression on cells from BM, spleen (SP), and LN. Representative density plots from each group are shown. (E) Representative images of hematoxylin-and-eosin (H&E)-stained BM, SP, liver, LN, and kidney sections; scale bar, 40 μm. Red arrow indicates an area with protein deposition mimicking the histologic findings in myeloma kidney. (F) Complete blood count (CBC) of peripheral blood samples collected from moribund mice and age-matched control mice. (G) Clonality analysis of VQ1 and VQ2 CD138+ cells using DJH recombination polymerase chain reaction (PCR) assay. *P < .05; **P < .01; ***P < .001. Hb, hemoglobin; I.P., intraperitoneal; ns, not significant; RBC, red blood cell; WBC, white blood cell.
A fraction of NrasQ61R/+; Vκ*MYC mice develop MM. (A) Acronyms of transgenic mouse lines and illustration of IgG1-Cre induction scheme. (B) SPEP was performed on representative mice from each group serially bled at the indicated weeks. The brackets show the γ-globulin component of the serum. (C) Kaplan-Meier survival curves (left) and disease incidences (right) of different groups of animals. MM is defined as CD138+B220− cells being >10% in BM, spleen, and/or LN along with end-organ abnormalities. (D) Flow cytometric analysis of B220 and CD138 expression on cells from BM, spleen (SP), and LN. Representative density plots from each group are shown. (E) Representative images of hematoxylin-and-eosin (H&E)-stained BM, SP, liver, LN, and kidney sections; scale bar, 40 μm. Red arrow indicates an area with protein deposition mimicking the histologic findings in myeloma kidney. (F) Complete blood count (CBC) of peripheral blood samples collected from moribund mice and age-matched control mice. (G) Clonality analysis of VQ1 and VQ2 CD138+ cells using DJH recombination polymerase chain reaction (PCR) assay. *P < .05; **P < .01; ***P < .001. Hb, hemoglobin; I.P., intraperitoneal; ns, not significant; RBC, red blood cell; WBC, white blood cell.
Based on our MM diagnostic criteria of >10% CD138+B220− plasma cells in any hematopoietic tissues (BM, spleen, and LN) plus evidence of end-organ damage, which is analogous to the International Myeloma Working Group (IMWG) criteria, only 1 of 13 V mice died with MM (Figure 1C; supplemental Table 2), though most of them developed an M spike at the moribund stage (Figure 1B). In addition, 11 of 13 V mice developed non-MM lymphoid malignancies (including B-cell lymphoma and non-MM plasmacytic diseases) based on presence of splenomegaly/lymphadenopathy, pathological evaluation, and flow cytometric analysis. The B cells from V mice with non-MM lymphoid malignancies were oligoclonal (supplemental Figure 3). One of 13 V mice died with a myeloproliferative neoplasm. The wide spectrum of malignancy phenotypes in V mice is not surprising, as the IgG1-Cre–mediated recombination of the floxed κ enhancer may perturb MYC expression to levels not optimal for myelomagenesis in the absence of a second molecular event (as corroborated by RNA-sequencing [RNA-Seq] analysis of the hMYC level seen in Figure 6A). We transplanted BM cells from 3 V mice into sublethally irradiated recipients (supplemental Figure 4). These donor mice developed either moderate myeloproliferative neoplasm (V1) or plasma cell disease (V2 and V3) at the moribund stage (supplemental Figure 4A-C). Currently, only 1 recipient developed an M spike and died after a prolonged latency (supplemental Figure 4D-E). Taken together, our data indicate that V mice demonstrate distinct pathological phenotypes from the reported Vκ*MYC mice in which the transgene is not subject to Cre recombination/rearrangement.
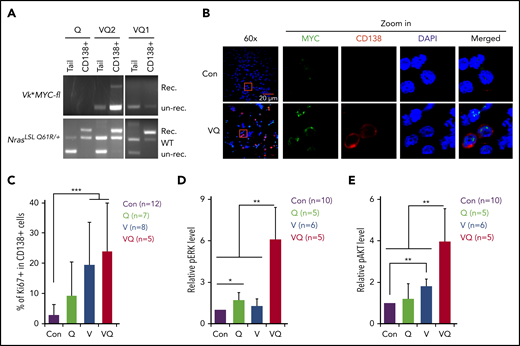
Molecular characterization of VQ myeloma cells. (A) Genotyping of NrasQ61R/+ and Vκ*MYC alleles to assess recombination efficiencies in VQ CD138+ cells. (B) hMYC expression in primary CD138+ plasma cells was detected using confocal immunofluorescence. Scale bar, 20 μm. The areas within the red rectangles were magnified (×5); original magnification ×60. (C) Quantification of Ki67+ cells in primary CD138+ cells from moribund VQ MM mice and age-matched control IgG1-Cre, V, and Q mice. (D-E) Quantification of phosphorylated ERK (pERK) (D) and pAKT levels (E) in primary CD138+ cells from moribund VQ MM mice and age-matched control IgG1-Cre, V, and Q mice. *P < .05; **P < .01; ***P < .001. DAPI, 4′,6-diamidino-2-phenylindole; Rec., recombined; un-rec., unrecombined.
Molecular characterization of VQ myeloma cells. (A) Genotyping of NrasQ61R/+ and Vκ*MYC alleles to assess recombination efficiencies in VQ CD138+ cells. (B) hMYC expression in primary CD138+ plasma cells was detected using confocal immunofluorescence. Scale bar, 20 μm. The areas within the red rectangles were magnified (×5); original magnification ×60. (C) Quantification of Ki67+ cells in primary CD138+ cells from moribund VQ MM mice and age-matched control IgG1-Cre, V, and Q mice. (D-E) Quantification of phosphorylated ERK (pERK) (D) and pAKT levels (E) in primary CD138+ cells from moribund VQ MM mice and age-matched control IgG1-Cre, V, and Q mice. *P < .05; **P < .01; ***P < .001. DAPI, 4′,6-diamidino-2-phenylindole; Rec., recombined; un-rec., unrecombined.
Of the Q mice, 1 of 7 died with MM and 6 of 7 died with either other lymphoid diseases (n = 3, including lymphoma and non-MM lymphoplasmacytic diseases) or without a defined hematologic malignancy (n = 3). The B cells in Q mice with other lymphoid diseases were also oligoclonal (supplemental Figure 3). We further transplanted BM or LN cells from 2 Q mice with a plasma cell disease (supplemental Figure 5A-C) into secondary recipients. The Q-recipient mice showed inconsistent M spikes (supplemental Figure 5D) and lacked bone lesions upon radiographic imaging (supplemental Figure 5E). They died within 15 months (supplemental Figure 5F). Despite moderate plasmacytosis in the LN (supplemental Figure 5G), we did not observe plasma cell infiltration to liver or kidney and no kidney abnormality was detected in the recipients (supplemental Figure 5H).
Both ERK and AKT pathways are hyperactivated in VQ myeloma cells
We characterized CD138+B220− cells from VQ myeloma mice, in particular VQ1 and VQ2. Genotyping analysis demonstrated that the NrasQ61R allele was fully activated in the myeloma cells, whereas the Vκ*MYC allele was predominantly unrecombined (Figure 2A). Expression of hMYC in myeloma cells was further validated using confocal immunofluorescence (Figure 2B). Unlike the indolent Vκ*MYC cells,8 V and VQ plasma cells were highly proliferative (Figure 2C). Similar to human advanced MM cells,32 both ERK and AKT pathways were hyperactivated in VQ MM cells (Figure 2D-E). By contrast, Q and V plasma cells only showed moderate hyperactivation of ERK and AKT, respectively.
VQ myeloma phenotypes are readily transplantable into syngeneic recipients
We transplanted VQ MM cells from multiple hematopoietic tissues into sublethally irradiated syngeneic recipients (Figure 3A; supplemental Figure 6B). VQ1 myeloma cells were enriched primarily in BM and secondarily in spleen, whereas VQ2 myeloma cells were localized primarily in LN and secondarily in BM and spleen (Figure 1D). Regardless of tissue origin, all of the transplant recipients developed an M spike (5-6 weeks for VQ-D1 recipients and 2-3 weeks for VQ-D2 recipients) (Figure 3B). VQ-D1 and VQ-D2 recipients died of malignant MM in 10 to 16 weeks and 3 to 5 weeks, respectively (Figure 3C). Our data indicate an extramedullary myeloma disease in VQ1 spleen and VQ2 LN. Similarly, VQ-D4 and VQ-D5 recipients died of malignant MM in 7 to 9 weeks and 6 to 7 weeks, respectively (supplemental Figure 6B). Clonality analysis showed that most of recipient mice had a dominant clone (supplemental Figure 6C). Radiographic imaging detected multiple osteolytic lesions in both VQ-D1 and VQ-D2 recipients (Figure 3D). Not surprisingly, all of them suffered from hind-limb paralysis at the moribund stage (Figure 3E). Flow cytometric analysis showed that VQ-D1 and VQ-D2 myeloma cells preserved their donor-characteristic tissue localization (Figure 3F). Similar to their donor mice, myeloma cell infiltration was observed in liver and kidney (Figure 3G). We did not detect surface expression of other B-cell markers (eg, CD19) (supplemental Figure 7A). Similar to their donor myeloma cells, VQ-D1 and VQ-D2 myeloma cells were hyperproliferative to a greater degree than the transplantable Vκ*MYC MM cells (∼35% in Chesi et al33 ) (supplemental Figure 7B). Associated with this high proliferation index, both ERK and AKT pathways were hyperactivated in VQ-D1 and VQ-D2 myeloma cells (supplemental Figure 7C).
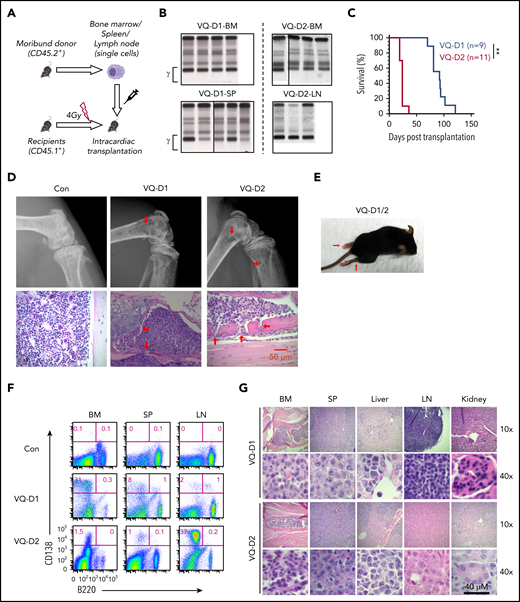
VQ myeloma is readily transplantable to syngeneic secondary recipients. (A) Schematic illustration of VQ myeloma transplantation strategy. (B) VQ1 BM cells (8 × 106; VQ-D1-BM), VQ1 splenocytes (20 × 106; VQ-D1-SP), VQ2 BM cells (5 × 106; VQ-D2-BM), or VQ2 LN cells (20 × 106; VQ-D2-LN) were transplanted into individual recipients. SPEP was performed on recipient mice bled at the moribund stage. The brackets show the γ-globulin component of the serum. The donor cells were isolated from BM, spleen (SP), or LN. (C) Kaplan-Meier survival curves of VQ-D1 and VQ-D2 recipient mice, which were transplanted with VQ1 and VQ2 myeloma cells, respectively. Log-rank test was performed. **P < .01. (D) Representative radiographic images and H&E-stained sections of hind-limb bones from moribund VQ-D1 and VQ-D2 recipient mice and age-matched control mice; scale bar, 50 μm. The red arrows indicate the osteolytic lesions. (E) Representative photo of VQ-D1/D2 recipients with hind-limb paralysis. (F) Flow cytometric analysis of B220 and CD138 expression on BM, SP, and LN cells from VQ-D1 and VQ-D2 recipient mice. Representative density plots are shown. (G) Representative images of H&E-stained BM, SP, liver, LN, and kidney sections; scale bar, 40 μm.
VQ myeloma is readily transplantable to syngeneic secondary recipients. (A) Schematic illustration of VQ myeloma transplantation strategy. (B) VQ1 BM cells (8 × 106; VQ-D1-BM), VQ1 splenocytes (20 × 106; VQ-D1-SP), VQ2 BM cells (5 × 106; VQ-D2-BM), or VQ2 LN cells (20 × 106; VQ-D2-LN) were transplanted into individual recipients. SPEP was performed on recipient mice bled at the moribund stage. The brackets show the γ-globulin component of the serum. The donor cells were isolated from BM, spleen (SP), or LN. (C) Kaplan-Meier survival curves of VQ-D1 and VQ-D2 recipient mice, which were transplanted with VQ1 and VQ2 myeloma cells, respectively. Log-rank test was performed. **P < .01. (D) Representative radiographic images and H&E-stained sections of hind-limb bones from moribund VQ-D1 and VQ-D2 recipient mice and age-matched control mice; scale bar, 50 μm. The red arrows indicate the osteolytic lesions. (E) Representative photo of VQ-D1/D2 recipients with hind-limb paralysis. (F) Flow cytometric analysis of B220 and CD138 expression on BM, SP, and LN cells from VQ-D1 and VQ-D2 recipient mice. Representative density plots are shown. (G) Representative images of H&E-stained BM, SP, liver, LN, and kidney sections; scale bar, 40 μm.
We continuously passaged VQ1 and VQ2 myeloma cells in vivo for 6 generations (supplemental Figure 8A). All of the VQ-D1 and VQ-D2 recipients developed an M spike at consistent time points and died of advanced MM as their corresponding donors. We also used cryopreserved myeloma cells for transplantation (supplemental Figure 8B). Again, all of the recipients developed MM-like phenotypes and died within a few weeks. We also transplanted cryopreserved VQ-D1 and -D2 cells into nonirradiated recipients (supplemental Figure 8C). Interestingly, 5 of 5 VQ-D1 recipients died with a highly malignant MM within 11 weeks, whereas only 1 of 4 VQ-D2 recipients died with MM upon observation for up to 20 weeks.
To identify which population(s) of cells contain myeloma-initiating activities, we first determined the donor contribution in CD138+B220−, CD138−B220+, and CD138−B220− cells (supplemental Figure 9A). As expected, >95% of CD138+B220− cells and >10% of CD138−B220− cells were donor-derived, whereas >99% of CD138−B220+ cells were derived from recipients (CD45.2−). Therefore, we fractionated VQ-D1 BM cells (supplemental Figure 9B) and VQ-D2 LN cells (supplemental Figure 9C) into CD138+B220− and CD138−B220− fractions and transplanted various numbers into sublethally irradiated recipients. Up to 3.5 million CD138−B220− cells failed to induce MM phenotypes (0 of 9), whereas as low as 500 CD138+B220− cells induced MM in 100% of recipients (10 of 10). We calculated that the frequency of myeloma-initiating cells was ∼1 of 150 to 1 of 200 of CD138+B220− cells, analogous to human myeloma.34
PD1 and TIGIT immune-checkpoint pathways are preserved in the VQ MM model
Programmed cell death protein 1 (PD1) and T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domain (TIGIT) are major immune-checkpoint pathways that mediate MM cell–induced T-cell suppression in MM patients.35-37 Therefore, we surveyed the expression of multiple immune-checkpoint ligands in primary VQ-D1 and VQ-D2 CD138+ MM cells. Our data showed that >95% of VQ-D1 MM cells and ∼59% of VQ-D2 MM cells expressed programmed death ligand 1 (PD-L1), and ∼50% of VQ-D1 MM cells and ∼18% of VQ-D2 MM cells expressed CD155 (Figure 4A). Expression of PD-L2 was not detectable on VQ-D1 and VQ-D2 MM cells. Moreover, >95% of VQ-D1 and VQ-D2 MM cells expressed major histocompatibility complex class I (MHC-I), but MHC class II (MHC-II) was only expressed in a few MM cells (Figure 4B). Corresponding to the PD-L1 and CD155 expression on MM cells, a fraction of CD8+ and CD4+ T cells in VQ-D1 and VQ-D2 recipient mice expressed the receptors, PD1 and TIGIT, respectively (Figure 4C-D). In VQ-D1 recipients, the T-cell compartment was only reduced in spleen, which was associated with a significant MM burden and increased PD1+ T cells (Figure 4E). In VQ-D2 recipients, both TIGIT and PD1 appeared to be expressed in a larger fraction of T cells than in control T cells (Figure 4D). Correspondingly, we observed a reduced T-cell compartment in the spleen and LN of VQ-D2 recipients (Figure 4F). Our data suggest that both PD1 and TIGIT pathways are preserved in VQ MM model.
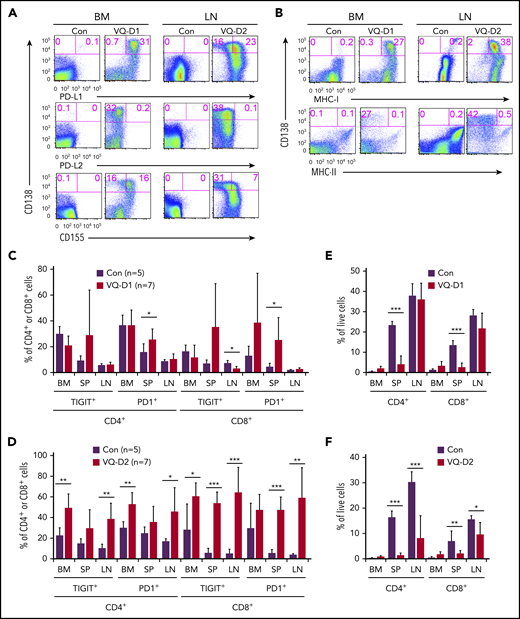
PD-L1/PD1 and CD155/TIGIT immune-checkpoint pathways are preserved in the VQ myeloma model. BM cells (1 × 106) from control (IgG1-Cre), VQ-D1, and VQ-D2 myeloma mice (>6 passages) were transplanted into sublethally irradiated recipients as described in Figure 3. (A-B) Flow cytometric analysis of CD138, PD-L1, PD-L2, CD155 (A), MHC-I, and MHC-II (B) on BM of VQ-D1 recipients and LN cells of VQ-D2 recipients. Representative density plots are shown. (C-D) Quantification of PD1+ and TIGIT+ T cells (CD8+ and CD4+) in BM, spleen (SP), and LN from age-matched control, VQ-D1 (C), and VQ-D2 (D) recipients. (E-F) Quantification of CD8+ and CD4+ T cells in BM, SP, and LN of control, VQ-D1 (E), and VQ-D2 (F) recipients. *P < .05; **P < .01; ***P < .001.
PD-L1/PD1 and CD155/TIGIT immune-checkpoint pathways are preserved in the VQ myeloma model. BM cells (1 × 106) from control (IgG1-Cre), VQ-D1, and VQ-D2 myeloma mice (>6 passages) were transplanted into sublethally irradiated recipients as described in Figure 3. (A-B) Flow cytometric analysis of CD138, PD-L1, PD-L2, CD155 (A), MHC-I, and MHC-II (B) on BM of VQ-D1 recipients and LN cells of VQ-D2 recipients. Representative density plots are shown. (C-D) Quantification of PD1+ and TIGIT+ T cells (CD8+ and CD4+) in BM, spleen (SP), and LN from age-matched control, VQ-D1 (C), and VQ-D2 (D) recipients. (E-F) Quantification of CD8+ and CD4+ T cells in BM, SP, and LN of control, VQ-D1 (E), and VQ-D2 (F) recipients. *P < .05; **P < .01; ***P < .001.
VQ myeloma cells display distinct transcriptional signatures from normal plasma cells and tVk12653 myeloma cells
We investigated the molecular mechanisms underlying VQ myeloma using single-cell RNA-Seq (scRNA-Seq) analysis. CD138+B220− cells were flow sorted from age-matched control BM and VQ-D2 BM and LN. Following removal of low-quality cells, a total of 749 single cells expressing >300 genes per cell were subjected to further analysis (supplemental Figure 10A-B). The median number of genes detected in normal plasma cells was ∼1000 and in VQ MM cells was ∼2000. Cluster analysis of all of these cells, visualized on a T-distributed stochastic neighbor embedding (tSNE) plot, yielded 5 distinct clusters (0-4) (Figure 5A; supplemental Table 3). Consistent with the previous report,38 normal plasma cells fell into 2 distinct clusters (C2 and C4) (Figure 5B). Based on the characteristic expression of genes associated with long-lived plasma cells (eg, Prg2 and Slpi) vs short-lived plasma cells (eg, CD93),39 C2 and C4 may represent long- and short-lived plasma cells, respectively. In contrast, the majority of VQ MM cells, regardless of their tissue origins, grouped into 3 clusters distinct from normal plasma cells (C0, C1, and C3), reflecting the intratumor heterogeneity. As in human MM patients,38 the minor populations of myeloma cells in C2 and C4 likely represent the residual normal plasma cells derived from the host recipient. As expected, normal plasma cells were nonproliferative and predominantly in G1 phase, whereas MM cells from BM and LN were highly proliferative and mostly in G2/M or S phases (Figure 5C). Gene-set enrichment analysis (GSEA) identified many pathways that were significantly enriched in VQ MM cells (P adjusted, <.25). Compared with normal plasma cells, VQ MM cells from BM were enriched for MYC and E2F target genes as well as genes involved in the unfolded protein-response pathway (Figure 5D). In addition, VQ MM cells showed significant enrichment of many human MM gene signatures associated with poor prognosis in human MM patients (Figure 5D-E),40 including the high-risk MM signature (UAMS-70) (supplemental Table 4) acknowledged by the IMWG at a significantly higher level than normal plasma cells.41 We also interrogated the high-risk MM gene signature, EMC-92.42 Unfortunately, more than one-half of the genes were not detected in control and VQ plasma cells. Interestingly, although VQ MM cells from BM and LN were largely clustered together, GSEA revealed that they displayed distinct transcriptional profiles in cell metabolism and epithelial-mesenchymal transition (supplemental Figure 10C).
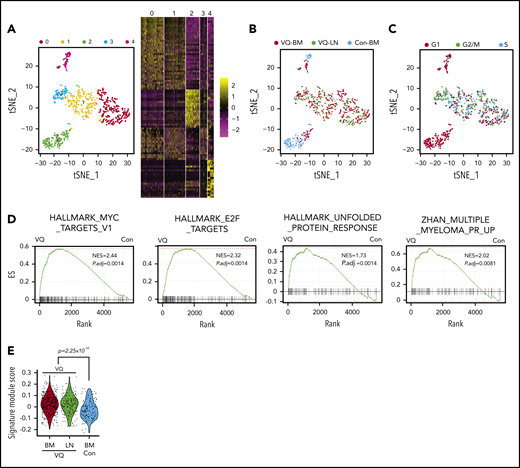
VQ myeloma cells display unique transcriptional signatures from normal plasma cells. CD138+B220− cells were sorted from age-matched control BM (Con-BM) and VQ-D2 BM and LN. scRNA-Seq analysis was performed as described in “Methods.” (A) tSNE plot depicting 749 single plasma cells derived from control and VQ-D2 mice. Each cluster is represented by a specific color and number. The top 20 genes expressed in each cluster are presented in the heatmap. (B) Samples are color-coded and projected on the tSNE plot (blue, Con-BM; red, VQ-BM; green, VQ-LN). (C) Cell-cycle phases are color-coded and projected on the tSNE plot (red, G1; green, G2/M; blue, S). (D) GSEA was performed between VQ-BM and Con-BM plasma cells. Adjusted P value (P.adj) and normalized enrichment score (NES) are shown on each plot. (E) Signature module scores were calculated for each cell based on the expression levels of genes associated with high-risk MM and the average expression profile of each cell. P value was determined using the Welch t test between VQ (BM + LN) and Con-BM. ES, enrichment score.
VQ myeloma cells display unique transcriptional signatures from normal plasma cells. CD138+B220− cells were sorted from age-matched control BM (Con-BM) and VQ-D2 BM and LN. scRNA-Seq analysis was performed as described in “Methods.” (A) tSNE plot depicting 749 single plasma cells derived from control and VQ-D2 mice. Each cluster is represented by a specific color and number. The top 20 genes expressed in each cluster are presented in the heatmap. (B) Samples are color-coded and projected on the tSNE plot (blue, Con-BM; red, VQ-BM; green, VQ-LN). (C) Cell-cycle phases are color-coded and projected on the tSNE plot (red, G1; green, G2/M; blue, S). (D) GSEA was performed between VQ-BM and Con-BM plasma cells. Adjusted P value (P.adj) and normalized enrichment score (NES) are shown on each plot. (E) Signature module scores were calculated for each cell based on the expression levels of genes associated with high-risk MM and the average expression profile of each cell. P value was determined using the Welch t test between VQ (BM + LN) and Con-BM. ES, enrichment score.
To validate our scRNA-Seq data, we performed bulk RNA-Seq using CD138+B220− BM cells sorted from wild-type (WT; n = 3), V (n = 3), tVk12653 (a transplantable Vκ*MYC line reported in Chesi et al33 ; n = 2), and VQ recipient mice (n = 5 from 3 donor lines) (Figure 6). Although Myc transcript levels in V (mean fragments per kilobase of transcript per million mapped reads [FPKM], 134) and VQ cells (mean FPKM, 279-579) were higher than that in WT cells (mean FPKM, 20), they were much lower than that in tVk12653 cells (mean FPKM, 2939) (Figure 6A). Notably, ∼99% of Myc transcripts in V, VQ, and tVk12653 cells are human MYC. Excluding immunoglobin genes, we found that the transcriptome of V cells was essentially identical as that of WT cells, whereas VQ and tVk12653 transcriptomes were distinct from the WT transcriptome (Figure 6B). Compared with WT cells, we identified a total of 3091 and 2654 genes differentially expressed in VQ and tVk12653 cells, respectively (false discovery rate, <0.05; fold change, >2) (Figure 6C). Consistent with our scRNA-Seq data, GSEA showed that MYC and E2F targets and genes involved in the unfolded-protein response pathway were enriched in VQ MM cells (Figure 6D). By contrast, only MYC and E2F targets were enriched in tVk12653 MM cells. Importantly, the UAMS-70 high-risk MM gene signature was significantly enriched in VQ cells but not in tVk12653 cells (Figure 6E), whereas the EMC-92 gene signature was not enriched in either type of MM cells (supplemental Figure 11). To understand the molecular differences of VQ vs tVk12653 MM cells, we directly compared their transcriptomes. We identified a total of 1217 genes differentially expressed in VQ cells (false discovery rate, <0.05; fold change, >2) (Figure 6F): 727 genes upregulated and 490 genes downregulated. Despite higher hMYC levels in tVk12653 MM cells, MYC target gene signatures were more significantly enriched in VQ cells (P adjusted, <.25) (Figure 6G), suggesting a prominent synergism between Nras Q61R signaling and hMYC. Consistent with our results previously described, E2F targets and genes involved in the unfolded-protein response pathway were also more enriched in VQ MM cells (P adjusted, <.25) (Figure 6G). These molecular changes associated with enrichment of RAS/ERK-signaling signatures in VQ cells (P adjusted, <.25) (Figure 6H). Our data demonstrate that Nras Q61R signaling plays an important role in driving high-risk VQ MM development.
![Transcriptomic profiling of normal plasma cells and MM models using bulk RNA-Seq. Bulk RNA-Seq analysis was performed using 50 000 sorted CD138+B220−CD45.2+ BM cells of WT (n = 3), V (n = 3), recipients of tVk12653 (n = 2), VQ-D1 (n = 2), VQ-D4 (n = 1), and VQ-D5 (n = 2). (A) Normalized expression levels of Myc in all samples. *P < .05; **P < .01; ***P < .001. (B) Scatterplots of average (avg) log10 FPKM values of all mapped genes of V, tVk12653, or VQ samples vs WT samples. Red and blue dots represent differentially upregulated and downregulated genes, respectively, for each comparison (log2 fold change [FC] >1 or <−1, adjusted P [P.adj], <.05); r2 value calculated from Pearson correlation coefficient. (C) Volcano plots illustrating the differentially expressed genes in tVk12653 or VQ MM cells vs WT plasma cells. GSEA was performed between tVk12653 or VQ and WT plasma cells against Hallmark (D) or UAMS-70 (E) gene sets. (F) Volcano plots illustrating the differentially expressed genes in VQ vs tVk12653 MM cells. GSEA analysis was performed between VQ and tVk12653 MM cells against Hallmark (G) or C2 curated (H) gene sets. Adjusted P value and normalized enrichment score (NES) are shown on each GSEA plot.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/1/10.1182_blood.2020007156/1/m_bloodbld2020007156f6.png?Expires=1769776644&Signature=u2vcT00XMLn53L5YWAcJBKUT303fABhyg9E3PuWf6A9brShsPFz92ilY4scfCU4WDSQZZsUIQYLUIRQgpcRVeVdRpG4mU7iSz-h5AZCGobk3XX~1V8J-PbdKSzEKRHjgm0R8LmmIuRiyM3wYqSFdcana24SXFOiDX~xrpKiGRY52qIdPxSKWpzx~Z3wZuHNC7UafGFp59g-mN~c3dGA6I2lVtOBSS9z2NMnemxtLteizjBYunG7dDvyBQpN4LCirYys8MXlQJ7gz4p9spQ6v6JRKZ8sSi5ZZ5AsJwMnVnhNGIREHX1KaBQNQJaE2HrAFao~NzRS4VVEFYZCKicJ5Kg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transcriptomic profiling of normal plasma cells and MM models using bulk RNA-Seq. Bulk RNA-Seq analysis was performed using 50 000 sorted CD138+B220−CD45.2+ BM cells of WT (n = 3), V (n = 3), recipients of tVk12653 (n = 2), VQ-D1 (n = 2), VQ-D4 (n = 1), and VQ-D5 (n = 2). (A) Normalized expression levels of Myc in all samples. *P < .05; **P < .01; ***P < .001. (B) Scatterplots of average (avg) log10 FPKM values of all mapped genes of V, tVk12653, or VQ samples vs WT samples. Red and blue dots represent differentially upregulated and downregulated genes, respectively, for each comparison (log2 fold change [FC] >1 or <−1, adjusted P [P.adj], <.05); r2 value calculated from Pearson correlation coefficient. (C) Volcano plots illustrating the differentially expressed genes in tVk12653 or VQ MM cells vs WT plasma cells. GSEA was performed between tVk12653 or VQ and WT plasma cells against Hallmark (D) or UAMS-70 (E) gene sets. (F) Volcano plots illustrating the differentially expressed genes in VQ vs tVk12653 MM cells. GSEA analysis was performed between VQ and tVk12653 MM cells against Hallmark (G) or C2 curated (H) gene sets. Adjusted P value and normalized enrichment score (NES) are shown on each GSEA plot.
Transcriptomic profiling of normal plasma cells and MM models using bulk RNA-Seq. Bulk RNA-Seq analysis was performed using 50 000 sorted CD138+B220−CD45.2+ BM cells of WT (n = 3), V (n = 3), recipients of tVk12653 (n = 2), VQ-D1 (n = 2), VQ-D4 (n = 1), and VQ-D5 (n = 2). (A) Normalized expression levels of Myc in all samples. *P < .05; **P < .01; ***P < .001. (B) Scatterplots of average (avg) log10 FPKM values of all mapped genes of V, tVk12653, or VQ samples vs WT samples. Red and blue dots represent differentially upregulated and downregulated genes, respectively, for each comparison (log2 fold change [FC] >1 or <−1, adjusted P [P.adj], <.05); r2 value calculated from Pearson correlation coefficient. (C) Volcano plots illustrating the differentially expressed genes in tVk12653 or VQ MM cells vs WT plasma cells. GSEA was performed between tVk12653 or VQ and WT plasma cells against Hallmark (D) or UAMS-70 (E) gene sets. (F) Volcano plots illustrating the differentially expressed genes in VQ vs tVk12653 MM cells. GSEA analysis was performed between VQ and tVk12653 MM cells against Hallmark (G) or C2 curated (H) gene sets. Adjusted P value and normalized enrichment score (NES) are shown on each GSEA plot.
VQ MM cell lines enable further genetic manipulations
To facilitate further genetic manipulations and drug screening/testing in the VQ MM model, we established 2 VQ MM cell lines, 4935 and 4938, from 2 independent VQ-D2 recipients (Figure 7A). Both cell lines showed MM-characteristic morphologies (Figure 7B) and displayed CD138+B220− immunophenotype (Figure 7C). As their donor VQ2 MM cells, the NrasQ61R allele was fully activated in both cell lines, whereas the Vκ*MYC allele was predominantly unrecombined (Figure 7D). hMYC was readily detectable in these cells (Figure 7E-F) and they divided approximately once per day. Above 95% of cells coexpressed PD-L1 and CD155 (Figure 7G). Approximately 80% of 4935 and 4938 cells expressed high levels of MHC-I but neither of the cell lines expressed MHC-II (Figure 7H). More importantly, both cell lines were easily infectible by retrovirus and lentivirus (Figure 7I), making further genetic modifications feasible in these cell lines.

Combined MEK inhibitor and pan bromodomain and extraterminal domain inhibitor ameliorate MM phenotypes and prolong the survival of VQ myeloma mice. (A-I) Two MM cell lines were established from VQ-D2 recipient mice. (A) Schematic illustration of the procedure to establish 2 VQ MM cell lines. (B) Representative images of H&E-stained 4935 and 4938 cells; original magnification ×100. (C) Flow cytometric assay of B220 and CD138 expression on the 2 cell lines. (D) Genotyping of NrasQ61R and Vκ*MYC alleles in the 2 cell lines. (E) hMYC expression in the cell lines was detected using confocal immunofluorescence. Scale bar, 10 μm. Middle and right panels: 4′,6-diamidino-2-phenylindole (DAPI) stain. (F) Hemagglutinin (HA)-tagged hMYC expression in the cell lines was validated using western blot analysis. Left, rabbit anti-Myc antibody; right, rat anti-HA antibody conjugated with horseradish peroxidase. (G-H) Flow cytometric analysis of CD138, PD-L1, CD155 (G), MHC-I, and MHC-II (H) expression on the cell lines. (I) Quantification of retroviral and lentiviral infection rate in the cell lines based on flow cytometric analysis of green fluorescent protein (GFP). (J-K) VQ MM cell lines were treated with increasing concentrations of trametinib for 48 hours. (J) Cell growth was measured using Cell TiterGlo assay. (K) Quantification of surface expression of PDL1 and CD155 in the presence of 10 nM trametinib. (L-M) VQ MM cell lines were treated with increasing concentrations of GSK525762 for 48 hours. (L) Cell growth was measured using Cell TiterGlo assay. (M) Quantification of surface expression of PDL1 and CD155 in the presence of 3 μM GSK525762. (N-O) VQ recipients were treated with vehicle or drug(s) as described in “Methods.” (N) SPEP was performed to quantify the γ/A ratios in VQ recipient mice before treatment (Pre) and at day 21 of treatment or at moribund before day 21 (Post). Note: some of the recipients were found dead and unable to be analyzed. Paired, 2-tailed Student t tests were performed. (O) Kaplan-Meier survival curves were plotted against days after treatment. Log-rank test was performed. *P < .05; **P < .01. G, GSK525762; IB, immunoblot; MFI, mean fluorescence intensity; MSCV, murine stem cell virus; T, trametinib; TG, trametinib and GSK525762; Veh, vehicle.
Combined MEK inhibitor and pan bromodomain and extraterminal domain inhibitor ameliorate MM phenotypes and prolong the survival of VQ myeloma mice. (A-I) Two MM cell lines were established from VQ-D2 recipient mice. (A) Schematic illustration of the procedure to establish 2 VQ MM cell lines. (B) Representative images of H&E-stained 4935 and 4938 cells; original magnification ×100. (C) Flow cytometric assay of B220 and CD138 expression on the 2 cell lines. (D) Genotyping of NrasQ61R and Vκ*MYC alleles in the 2 cell lines. (E) hMYC expression in the cell lines was detected using confocal immunofluorescence. Scale bar, 10 μm. Middle and right panels: 4′,6-diamidino-2-phenylindole (DAPI) stain. (F) Hemagglutinin (HA)-tagged hMYC expression in the cell lines was validated using western blot analysis. Left, rabbit anti-Myc antibody; right, rat anti-HA antibody conjugated with horseradish peroxidase. (G-H) Flow cytometric analysis of CD138, PD-L1, CD155 (G), MHC-I, and MHC-II (H) expression on the cell lines. (I) Quantification of retroviral and lentiviral infection rate in the cell lines based on flow cytometric analysis of green fluorescent protein (GFP). (J-K) VQ MM cell lines were treated with increasing concentrations of trametinib for 48 hours. (J) Cell growth was measured using Cell TiterGlo assay. (K) Quantification of surface expression of PDL1 and CD155 in the presence of 10 nM trametinib. (L-M) VQ MM cell lines were treated with increasing concentrations of GSK525762 for 48 hours. (L) Cell growth was measured using Cell TiterGlo assay. (M) Quantification of surface expression of PDL1 and CD155 in the presence of 3 μM GSK525762. (N-O) VQ recipients were treated with vehicle or drug(s) as described in “Methods.” (N) SPEP was performed to quantify the γ/A ratios in VQ recipient mice before treatment (Pre) and at day 21 of treatment or at moribund before day 21 (Post). Note: some of the recipients were found dead and unable to be analyzed. Paired, 2-tailed Student t tests were performed. (O) Kaplan-Meier survival curves were plotted against days after treatment. Log-rank test was performed. *P < .05; **P < .01. G, GSK525762; IB, immunoblot; MFI, mean fluorescence intensity; MSCV, murine stem cell virus; T, trametinib; TG, trametinib and GSK525762; Veh, vehicle.
MEK inhibition–based combination therapies attenuate MM phenotypes and prolong the survival of VQ recipients
We further tested whether the VQ MM model can be used as a preclinical platform for evaluating established and potentially novel therapeutic agents. We treated VQ MM cell lines with AZD6244 (a potent MEK inhibitor28 ) (supplemental Figure 12A) or bortezomib (a proteasome inhibitor commonly used in human MM treatment) (supplemental Figure 12B). Both agents induced a dose-dependent inhibition of MM cell growth with 50% inhibitory concentration (IC50) at 1 to 10 μM (AZD6244) and 15 to 20 nM (bortezomib). The IC50 of bortezomib in VQ MM cell lines is notably higher than its IC50 for growth inhibition of bortezomib-sensitive human MM cell lines (<10 nM),43 suggesting that VQ MM cells may show de novo resistance to bortezomib. To determine the effects of AZD6244 and bortezomib on MM development in vivo, we transplanted primary VQ-D1 MM cells into sublethally irradiated recipients (supplemental Figure 12C). Once advanced MM was established as indicated by a high γ-globulin/albumin (γ/A) ratio (average, 0.6-0.8), the mice were divided into 4 groups with comparable ratios and treated with vehicle, bortezomib alone, AZD6244 alone, or combined bortezomib and AZD6244 for 21 days. SPEP analysis was performed at day 28 or at a moribund stage before day 28. Our data showed that compared with vehicle-treated mice, combinatorial treatment of AZD6244 and bortezomib ameliorated MM phenotypes, including stabilizing the γ/A ratio (average, 0.7) (supplemental Figure 12D), and elevated red blood cell and platelet counts (supplemental Figure 12E). Moreover, bortezomib/AZD6244 treatment prolonged the survival of VQ recipients (supplemental Figure 12F). The effects of combination therapy tended to be better than single agent alone.
Encouraged by these results, we switched to trametinib, the MEK inhibitor approved by the US Food and Drug Administration (FDA) for treating melanoma.44 Trametinib displayed a more potent inhibition of VQ MM cell growth in vitro (IC50, 5-10 nM) (Figure 7J). Moreover, upon trametinib treatment, surface expression of PD-L1 was significantly reduced whereas CD155 expression was not changed (Figure 7K). Because targeting MYC via bromodomain inhibition confers significant antimyeloma benefit,9 we also tested GSK525762, a pan bromodomain and extraterminal domain inhibitor currently under clinical development.45,46 GSK525762 showed a moderate inhibition of MM cell growth in vitro (IC50, ∼3 μM) (Figure 7L) and had no effects on PD-L1 and CD155 surface expression in MM cells (Figure 7M). Given a much better antileukemia effect of GSK525762 in vivo than in vitro (X.Y., unpublished data), we moved forward to the in vivo study. We transplanted primary VQ-D1 MM cells into sublethally irradiated recipients. Upon development of an advanced MM characterized by a high γ/A ratio (average, ∼0.5) (Figure 7N), the mice were divided into 4 groups as previously described and treated with vehicle, trametinib, GSK525762, or combined trametinib and GSK525762 until a moribund stage. SPEP analysis was performed at day 21 or at a moribund stage before day 21. Vehicle- or GSK525762-treated mice died within 6 weeks, whereas trametinib treatment significantly prolonged the survival of diseased mice (Figure 7O). Combinatorial drug treatment outperformed both single-agent treatments (Figure 7N-O). Our results corroborate the clinical success of using trametinib in combination therapy regimens for some patients with advanced MM,47 suggesting that the VQ MM model may serve as a robust platform for preclinical studies of therapeutic agents with novel modes of action.
Discussion
Activating mutations of RAS-pathway genes constitute the most frequent genetic event in advanced and refractory MM. Here, we describe the first genetically engineered Ras; MYC-driven model for advanced MM. This model displays many biological and clinical features characteristic for human advanced MM and thus provides a platform to investigate MM progression in vivo and evaluate potential therapeutic agents.
KRAS mutations vs NRAS mutations in MM
Despite the high degree of sequence identity between Kras and Nras proteins (>80%), we and others observed that oncogenic Kras is a much more potent oncogene than oncogenic Nras in myeloid diseases, demonstrated by earlier disease onset, more severe phenotypes, and shorter survival in KrasG12D/+ mice compared with NrasG12D/+, NrasG12D/G12D, and NrasQ61R/+ mice (reviewed in Chang et al48 ). Interestingly, when activated throughout the hematopoietic system, KrasG12D/+ mice never developed B-cell malignancies whereas a fraction of oncogenic Nras mice did.22,28,49 One possibility could be that strong oncogenic Kras signaling is less favorable than oncogenic Nras in B cells. In human MM, KRAS and NRAS mutation frequencies are approximately equal.11,13,14 However, NRAS mutations occur in untreated advanced patients and relapsed patients at a 50:50 ratio, whereas ∼2 of 3 KRAS mutations are acquired in relapsed patients, suggesting that NRAS signaling may be equally important in MM progression and drug resistance whereas KRAS signaling may be more involved in drug resistance.
Although mutations at codons G12, G13, and Q61 of RAS are all perceived oncogenic equivalents, they display distinct biochemical and functional properties. RAS G12 and G13 mutations affect the GTPase activity of RAS protein by preventing proper position of the RAS-GTPase activation protein arginine finger within the catalytic site, whereas Q61 mutations lead to more severe impairment of RAS-intrinsic GTPase function.50 Therefore, G12 and G13 mutations are less activating than Q61 mutations. Not surprisingly, we and others found that the oncogenic activity of Nras is codon-dependent.16,27 Corroborating the mouse results, in human hematologic malignancies with NRAS mutations, G12 mutations are prevalent in myeloid leukemia, whereas Q61 mutations are predominant in MM (∼75%).27 In contrast, the codon preference in the KRAS gene is much less prominent. In MM patients with KRAS mutations, both codons G12 and Q61 are hot spots with an equal mutation rate based on the Multiple Myeloma Research Foundation (MMRF) database (G12, 39%; Q61, 37%) and the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (G12, 32%; Q61, 36%). Equal mutation rates on KRAS G12 and Q61 may be due to the unique capability of KrasG12D, but not oncogenic Nras, to cross-activate WT Ras via Sos1, a Ras guanine nucleotide exchange factor,51 leading to a more potent hyperactivation of MEK/ERK than KrasG12D alone. This process could minimize the functional difference between KRAS G12 and Q61 mutations.
Functional contribution of NrasQ61R to the VQ model
Compared with the de novo Vκ*MYC model, which reflects a more indolent, less proliferative MM, the VQ MM model shares similar developmental origins but displays distinct and more diversified antibody isotypes (Chesi et al8 ; supplemental Table 1); VQ cells display several non-IgM isotypes, whereas Vκ*MYC cells are predominantly of IgG1 isotype. Of note, in all 3 VQ mice with both IgG and IgA clones, the IgA clone became dominant upon transplantation and VQ-D2 cell lines were also derived from the IgA clone (supplemental Table 1). The various isotypes in the VQ model may reflect the demonstrated expression of Cre recombinase in non-IgG–switched plasma cells from the IgG-Cre transgene.18 VQ survival is significantly shorter than that of de novo Vκ*MYC mice (Figure 3C), whereas VQ transplant recipients have a comparable survival to that of selected bortezomib-resistant Vκ*MYC lines.33 In contrast to Vκ*MYC and Vκ*MYC-derived transplantable lines, VQ2-derived MM cells were readily propagated in vitro and were efficiently infectible by standard lentiviral and retroviral vectors (Figure 7A-C).
We believe that NrasQ61R expression drives progression from the indolent Vκ*MYC model to the advanced, high-risk VQ model through hyperactivation of the MEK/ERK pathway (Kong et al27 ; Figure 2D; supplemental Figure 7C), promoting MM hyperproliferation (Figure 2C; supplemental Figure 7B), upregulating MYC and E2F target genes (Figures 5D and 6D,G) and PD-L1 (Figures 4A and 7M), and resulting in more resistance to bortezomib than the original Vκ*MYC mice33 (supplemental Figure 12B,F). Consistent with this idea, NRAS Q61 mutations are significantly associated with bortezomib resistance in human MM patients.42
The single-cell and bulk RNA-seq data have been deposited in the Gene Expression Omnibus database and made available to the public (accession number GSE153628).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the University of Wisconsin Carbone Comprehensive Cancer Center (UWCCC) for use of its Shared Services (Flow Cytometry Laboratory, Genome Editing and Animal Models Shared Resource, and Experimental Pathology Laboratory) to complete this research.
The Flow Cytometry Laboratory was supported by National Institutes of Health (NIH) Shared Instrument Grants 1S10RR025483-01 (from the National Institute of General Medical Sciences for the BD FACS Aria II BSL-2 Cell Sorter) and 1S100OD018202-01 (from the Office of the Director for the BD LSR Fortessa). This work was supported by NIH grants T32 GM081061 (E.D.F.) from the National Institute of General Medical Sciences; AI079087 (D.W.) from the National Institute of Allergy and Infectious Diseases; HL130724 (D.W.) from the National Heart, Lung, and Blood Institute; and R01CA152108 (J.Z.) from the National Cancer Institute. This work was also supported by a Scholar Award from the Leukemia & Lymphoma Society (J.Z.), a Translational Research Program grant (6551-18) from the Leukemia & Lymphoma Society, and a Research Scholar Award (127508-RSG-15-045-01-LIB) from the American Cancer Society (F.A.). This work was supported, in part, by NIH National Cancer Institute grant P30 CA014520 for UW Comprehensive Cancer Center (UWCCC) support and the UWCCC Trillium Fund for Myeloma Research.
Authorship
Contribution: Z.W., A.R., D.W., F.A., and J.Z. conceived and designed the study; Z.W., A.R., G.Y., E.D.F., Z.T.M., A.P., and R.F. acquired data; Z.W., A.R., G.Y., M.C., E.D.F., Z.T.M., A.P., E.A.R., Q.F., R.T.B., P.L.B., D.W., F.A., and J.Z. analyzed and interpreted data; A.C.P., S.L., Y.Z., X.Y., J.J., M.Y., A.C., and J.M. provided technical or material support; Z.W., A.R., G.Y., E.D.F., Z.T.M., A.P., E.A.R., C.S.M., N.S.C., P.L.B., D.W., F.A., and J.Z. wrote, reviewed, and revised the manuscript; and F.A. and J.Z. supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Fotis Asimakopoulos, UCSD Moores Cancer Center, MCC2308, Mailbox 0960, 3855 Health Sciences Dr, La Jolla, CA 92093; e-mail: fotis@ucsd.edu; or Jing Zhang, McArdle Laboratory for Cancer Research, University of Wisconsin-Madison, Room 7453, WIMR II,1111 Highland Ave, Madison, WI 53705; e-mail: zhang@oncology.wisc.edu.
