

Key Points
Single-cell analyses determine the trajectory from endothelial cells to pre-HSCs, with defined intermediate stages.
Hemogenic endothelial cells in the arteries produce 2 waves of CD45+ cells; an early wave of progenitors followed by pre-HSCs.

Abstract
Hematopoietic stem and progenitor cells (HSPCs) in the bone marrow are derived from a small population of hemogenic endothelial (HE) cells located in the major arteries of the mammalian embryo. HE cells undergo an endothelial to hematopoietic cell transition, giving rise to HSPCs that accumulate in intra-arterial clusters (IAC) before colonizing the fetal liver. To examine the cell and molecular transitions between endothelial (E), HE, and IAC cells, and the heterogeneity of HSPCs within IACs, we profiled ∼40 000 cells from the caudal arteries (dorsal aorta, umbilical, vitelline) of 9.5 days post coitus (dpc) to 11.5 dpc mouse embryos by single-cell RNA sequencing and single-cell assay for transposase-accessible chromatin sequencing. We identified a continuous developmental trajectory from E to HE to IAC cells, with identifiable intermediate stages. The intermediate stage most proximal to HE, which we term pre-HE, is characterized by increased accessibility of chromatin enriched for SOX, FOX, GATA, and SMAD motifs. A developmental bottleneck separates pre-HE from HE, with RUNX1 dosage regulating the efficiency of the pre-HE to HE transition. A distal candidate Runx1 enhancer exhibits high chromatin accessibility specifically in pre-HE cells at the bottleneck, but loses accessibility thereafter. Distinct developmental trajectories within IAC cells result in 2 populations of CD45+ HSPCs; an initial wave of lymphomyeloid-biased progenitors, followed by precursors of hematopoietic stem cells (pre-HSCs). This multiomics single-cell atlas significantly expands our understanding of pre-HSC ontogeny.
Introduction
Hematopoietic ontogeny involves multiple “waves” in which hematopoietic stem and progenitor cells (HSPCs) with different potentials differentiate from hemogenic endothelial (HE) cells. HE cells in the yolk sac (YS) differentiate into committed erythromyeloid progenitors (EMP) and lymphoid progenitors, and the caudal arteries produce lymphoid progenitors and prehematopoietic stem cells (HSCs).1,2 YS hematopoiesis can be recapitulated in embryonic stem (ES) cell cultures, where the molecular events are well-described.3,4 Groundbreaking studies described the transcriptomes of HE and pre-HSCs in the major caudal artery, the dorsal aorta, at single-cell resolution.5-8 However, these analyses did not examine the distribution or chromatin landscapes of cells along the trajectory, or the heterogeneity of cells in the intra-arterial clusters (IACs), because of the limited number of cells sequenced. To gain insights into the molecular mechanisms mediating the differentiation of arterial endothelial (E) cells into IACs we used the 10x Genomics platform and performed single-cell RNA sequencing (scRNA-Seq) and single-cell assay for transposase-accessible chromatin sequencing (scATAC-Seq). Our data reveal a continuous trajectory from E to IAC cells, previously undefined transitional cell populations along the trajectory, the pathways and transcription factors active in these cells, and describe the molecular heterogeneity of IAC cells.
Methods
Animal husbandry
B6C3F1/J 3-week-old female mice were purchased from The Jackson Laboratory (stock no. 100010). Females were injected with 5 IU pregnant mare serum gonadotropin and 48 hours later with 5 IU human chorionic gonadotropin (MilliporeSigma), then immediately paired overnight with C57BL6/J male mice. Runx1:GFP (Runx1tm4Dow)9 homozygous male mice were mated to superovulated B6C3F1/J 3-week-old female mice to generate embryos for purification of E and HE cells. Female B6C3F1/J mice were mated with male B6129SF1/J mice for isolating fetal liver HSCs. Ectopic RUNX1 expression in E cells in Tg(Cdh5-cre/ERT2)1Rha embryos10 that contained an activatable Runx1 complementary DNA in the Rosa26 locus was described previously.11 Runx1+/− mice (Runx1tm1Spe) were described previously.12 The morning after mating is considered 0.5 days post coitus (dpc). Embryos 9.5 to 11.5 dpc were accurately staged at the time of harvest by counting somites. Embryos that showed abnormal 2wdevelopment were discarded. Mice were handled according to protocols approved by the University of Pennsylvania’s Institutional Animal Care and Use Committee and housed in a specific pathogen-free facility.
scRNA-Seq
Sorted cells were immediately processed for library preparation using the 10x Genomics Chromium Single Cell 3′ Reagent Kit v.2. Indexed libraries were pooled and sequenced on an Illumina HiSeq 4000 or NextSeq 550 using paired-end 26 × 98 bp read length.
scATAC-Seq
Sorted cells were processed to prepare single nuclei suspension. Nuclei suspension was processed for library preparation using the Chromium Single Cell Assay for Transposase-Accessible Chromatin (ATAC) Reagent Kit's protocol. Indexed libraries were pooled and sequenced on an Illumina NextSeq 550 using paired-end 50 × 50 bp read length.
See supplemental Data on the Blood Web site for additional description of methods and materials.
Results
scRNA-Seq reveals a continuous trajectory from E to IAC cells
Our strategy was to analyze all cells along the trajectory in a single sample to determine their distribution between different transcriptional states and combine that with analyses of purified subpopulations to make accurate cell assignments and obtain additional coverage of rare cells (Figure 1A). We captured the entire trajectory by purifying a population containing all E, HE, and IAC cells (E+HE+IAC) from 9.5 to 10.5 dpc embryos using a combination of E markers (supplemental Figure 1A). We also purified subpopulations of HE and E cells from 9.5 and 10.5 dpc embryos based on expression of green fluorescent protein (GFP) from the Runx1 locus9 (supplemental Figure 1B). We confirmed that only HE cells were capable of producing hematopoietic cells ex vivo (supplemental Figure 1C). Kithi IAC cells were excluded in the sorts, therefore HE and E cells were negligibly contaminated with HSPCs (supplemental Figure 1D). We purified IAC cells from 10.5 and 11.5 dpc embryos using antibodies recognizing E markers and Kit, 9.5 dpc yolk sac EMPs (YS-EMPs), and 14.5 dpc fetal liver HSCs (FL-HSCs) (supplemental Figure 2).
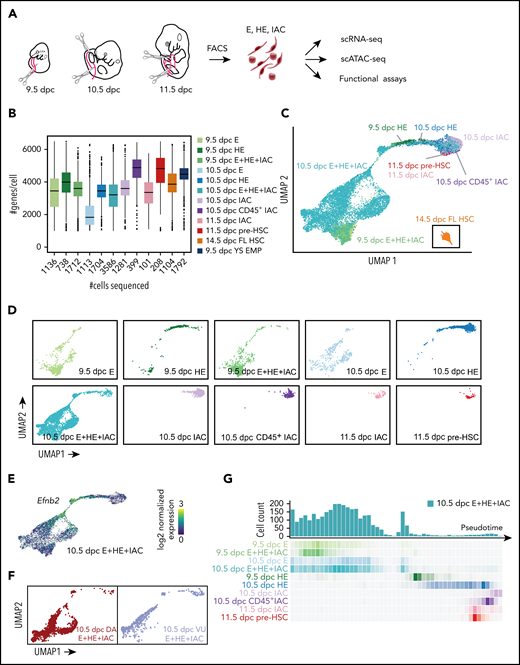
Experimental design and overview of single cell RNA-Seq data. (A) The caudal part of embryos was isolated (boundaries are illustrated with scissors), and then organs and gut tube removed. VU were isolated and included in the sample. The tissue was dissociated and cells were isolated by fluorescence-activated cell sorting, then analyzed by scRNA-Seq, scATAC-Seq, or in functional assays. All cell populations purified and sequenced are listed in supplemental Table 2; sort plots are shown in supplemental Figures 1 and 2. (B) The number of cells sequenced (x-axis) and genes per cell detected for representative samples. (C) UMAP of continuous EHT trajectory and FL-HSCs, with selected cell populations labeled. (D) Distribution of cells from each dataset in the UMAP reflecting EHT trajectory. (E) UMAP illustrating the 2 streams of E cells expressing high levels of the arterial marker Efnb2 that converge to form the stem leading to HE and IACs. (F) E+HE+IAC cells separately purified from the VU arteries, and from the DA within the caudal half of the embryo, highlighted on the global UMAP plot. (G) Cell count along the pseudotime trajectory. Bar graph quantifies results from a single sort of 10.5 dpc E+HE+IAC cells; heat maps below the graph show distribution of cells in all sorted cell populations.
Experimental design and overview of single cell RNA-Seq data. (A) The caudal part of embryos was isolated (boundaries are illustrated with scissors), and then organs and gut tube removed. VU were isolated and included in the sample. The tissue was dissociated and cells were isolated by fluorescence-activated cell sorting, then analyzed by scRNA-Seq, scATAC-Seq, or in functional assays. All cell populations purified and sequenced are listed in supplemental Table 2; sort plots are shown in supplemental Figures 1 and 2. (B) The number of cells sequenced (x-axis) and genes per cell detected for representative samples. (C) UMAP of continuous EHT trajectory and FL-HSCs, with selected cell populations labeled. (D) Distribution of cells from each dataset in the UMAP reflecting EHT trajectory. (E) UMAP illustrating the 2 streams of E cells expressing high levels of the arterial marker Efnb2 that converge to form the stem leading to HE and IACs. (F) E+HE+IAC cells separately purified from the VU arteries, and from the DA within the caudal half of the embryo, highlighted on the global UMAP plot. (G) Cell count along the pseudotime trajectory. Bar graph quantifies results from a single sort of 10.5 dpc E+HE+IAC cells; heat maps below the graph show distribution of cells in all sorted cell populations.
Summary statistics for collected cell populations are in Figure 1B and supplemental Table 2. We used uniform manifold approximation and projection (UMAP) to reduce the data dimension.14 After filtering out non-E and nonhematopoietic cells (supplemental Figure 3A) and reducing batch effect using an “informative feature selection” method (supplemental Figure 4; supplemental Methods), UMAP of the combined datasets shows a continuous trajectory from E to IAC cells (Figure 1C-D; supplemental Figure 3). 14.5 dpc FL-HSCs are disconnected from this trajectory, therefore, are more distantly related (Figure 1C).
Two streams of Efbn2+ E cells in the UMAP converge to form a stem leading to HE and IACs (Figure 1E). Analyses of 10.5 dpc E+HE+IAC cells manually separated into Vitelline and umbilical (VU) arteries and dorsal aorta (DA) demonstrated that VU cells contribute to one of these streams and DA to both streams (Figure 1F). The E+HE+IAC samples, which demonstrate the distribution of cells at various stages, show that at 9.5 dpc, IAC cells constitute only 0.5% of the E+HE+IAC population, but at 10.5 dpc the fraction of IAC cells expands sevenfold, representing 3.5% of the population, consistent with histological analyses showing increased numbers of IACs between these 2 embryonic stages15,16 (Figure 1D,G).
Unsupervised clustering identified 7 distinct populations in the combined dataset, and separated the 2 streams of E cells into distinct clusters (supplemental Figure 5A-C). One cluster, containing only DA E cells, expresses high levels of Wnt target genes (Wnthi E) (supplemental Figure 5D). The second cluster, Wntlo E containing both DA and UV cells, expresses lower levels of Wnt target genes. Wnthi E and Wntlo E could be further subdivided into arterial E (AE) and venous E (VE) by computing an arterial/venous score based on sets of AE- and VE-specific genes17 (Figure 2A-D). Pseudotime-ordered Wnthi and Wntlo E cells before the point where the AE score exceeded the VE score were defined as VE, and after that point were defined as AE. Wnthi AE and Wntlo AE then converge to form a distinct cluster determined by both UMAP and a recent visualization method PHATE18 that we termed conflux AE (Figure 2A; supplemental Figure 5E). The confluence of transcriptomes in conflux AE is driven by the loss of Wnthi/lo AE-specific gene expression and increased levels of transcripts from later stage-specific genes (Figure 2D-E). For example, expression of Wnt target genes Foxq1 and Nkd1 in Wnthi AE cells, and Tmem255a in Wntlo AE cells are downregulated in conflux AE (Figure 2D; supplemental Table 3). Cell cycle is also significantly inhibited in conflux AE (supplemental Figure 5F-G), whereas Notch signaling is elevated, seen by increased expression of the Notch ligand Dll4 and transcription factor Hey2 (Figure 2D; supplemental Figure 3C). Additional pathways activated in conflux AE include those regulating cell shape and motility (“elastin fiber formation,” “platelet adhesion to exposed collagen,” “gap junction assembly”) and processes important in hematopoietic cells (“MAPK signaling for integrins”) (Figure 2F).
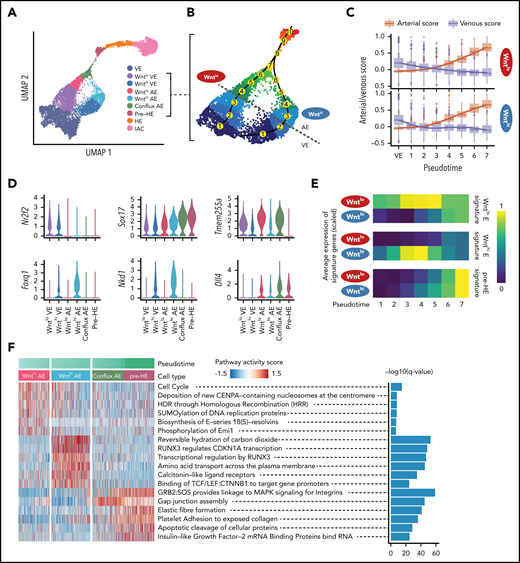
Two streams of E cells converge before hemogenic endothelium. (A) UMAP of EHT trajectory (from Figure 1C, with FL-HSC removed) showing the 7 clusters identified by Louvain clustering in supplemental Figure 5A, with Wnthi E subdivided into Wnthi AE and Wnthi VE, plus Wntlo E subdivided into Wntlo AE and Wntlo VE based on the arterial/venous score determined as shown in panels B and C. (B) Enlarged UMAP highlighting the 2 streams of endothelial cells converging to conflux AE. Numbers in yellow circles represent pseudotime bins up to the point of convergence. The dotted gray line represents the boundary between AE and VE. (C) Arterial score vs venous score over pseudotime bins. Cluster VE from panel A is used as the first pseudotime bin. Curves are fitted for AE score and VE score of each branch using a generalized additive model. (D) Violin plots of expression of cluster-specific genes, including venous marker Nr2f2, arterial marker Sox17, Wntlo AE-specific gene Tmem255a, Wnthi AE-specific genes Foxq1 and Nkd1, and Notch ligand Dll4. (E) Average expression of Wntlo E, Wnthi E, and pre-HE-specific genes over pseudotime. Differentially expressed genes were derived by pairwise expression analysis between Wntlo E and Wnthi E. Pre-HE specific genes were derived by comparing pre-HE with Wntlo plus Wnthi E. (F) Heatmap showing stream-specific Reactome pathway activity over pseudotime. AUCell package63 was used to compute a pathway activity score for each cell. One vs the rest Student t test was used to identify group-specific pathways and the top 6 most significant pathways were plotted.
Two streams of E cells converge before hemogenic endothelium. (A) UMAP of EHT trajectory (from Figure 1C, with FL-HSC removed) showing the 7 clusters identified by Louvain clustering in supplemental Figure 5A, with Wnthi E subdivided into Wnthi AE and Wnthi VE, plus Wntlo E subdivided into Wntlo AE and Wntlo VE based on the arterial/venous score determined as shown in panels B and C. (B) Enlarged UMAP highlighting the 2 streams of endothelial cells converging to conflux AE. Numbers in yellow circles represent pseudotime bins up to the point of convergence. The dotted gray line represents the boundary between AE and VE. (C) Arterial score vs venous score over pseudotime bins. Cluster VE from panel A is used as the first pseudotime bin. Curves are fitted for AE score and VE score of each branch using a generalized additive model. (D) Violin plots of expression of cluster-specific genes, including venous marker Nr2f2, arterial marker Sox17, Wntlo AE-specific gene Tmem255a, Wnthi AE-specific genes Foxq1 and Nkd1, and Notch ligand Dll4. (E) Average expression of Wntlo E, Wnthi E, and pre-HE-specific genes over pseudotime. Differentially expressed genes were derived by pairwise expression analysis between Wntlo E and Wnthi E. Pre-HE specific genes were derived by comparing pre-HE with Wntlo plus Wnthi E. (F) Heatmap showing stream-specific Reactome pathway activity over pseudotime. AUCell package63 was used to compute a pathway activity score for each cell. One vs the rest Student t test was used to identify group-specific pathways and the top 6 most significant pathways were plotted.
Runx1 regulates progression through a developmental bottleneck between pre-HE and HE
Conflux AE gives rise to HE and IAC cells, which are characterized by high levels of Runx1 and Gfi1 and IAC by expression of the pan-hematopoietic marker gene Ptprc (encoding CD45) (Figure 3B; supplemental Figure 3C). Between conflux AE and HE is a distinct cluster of E cells that we named pre-HE. UMAP and pseudotime trajectories of E10.5 E+HE+IAC reveal an accumulation of pre-HE cells, suggesting a bottleneck between pre-HE and HE (Figure 3C) that is prominent at 10.5 dpc although not at 9.5 dpc (Figure 1D). Gfi1, a direct RUNX1 target that participates in extinguishing E fate,19 shows elevated expression immediately after cells pass through the bottleneck and become HE, whereas high levels of Sox17, the Notch target Hey2, and the arterial marker Cd44 are found in prebottleneck populations including conflux E and pre-HE (Figure 3B). To provide further evidence for the bottleneck, we used Velocyto and scVelo, which infer directionality of differentiation by modeling dynamics of unspliced vs spliced RNAs when a gene is up- or downregulated.20,21 Velocyto and scVelo showed a marked decrease in RNA velocity in pre-HE cells, suggesting a differentiation barrier restricting their progression toward HE (Figure 3C; supplemental Figure 6). Once pre-HE cells transit to HE, however, they smoothly differentiate to IAC cells. Several pathways known to promote Runx1 expression and HSPC formation are upregulated in pre-HE, including Notch, tumor necrosis factor, fluid shear stress, cytokine signaling, and synthesis of eicosanoids, vitamins, and sterols,22-29 suggesting these pathways are important in pre-HE (Figure 3D; supplemental Figure 7; supplemental Table 4). Once cells transition to HE, RUNX1 plays a predominant role.
![Developmental bottleneck between pre-HE and HE cells. (A) UMAP of E10.5 E+HE+IAC cells showing 9 cell types from Figure 2A. (B) Expression of key markers of clusters, including Hey2 in conflux AE and pre-HE, Cd44 in conflux AE, pre-HE, HE, and IACs; Ptprc in IACs; Gfi1 and Runx1 in HE and IACs; and high levels of Sox17 in conflux AE and pre-HE, with downregulation in HE. Note Runx1 is expressed at low levels in all subsets of endothelial cells. (C) Velocyto analysis revealing different differentiation dynamics along the EHT in ED10.5 E+HE+IAC cells. To the right is an enlargement velocity of pre-HE cells that have accumulated at the bottleneck between pre-HE and HE. (D) Activity of pathways from Kyoto Encyclopedia of Genes and Genomes database, computed for each cell using the AUCell method.63 (E) UMAP of E+HE+IAC cells from 10.5 dpc Runx1+/+ and Runx1+/− littermates. Bars at bottom depict the distribution of cells between conflux AE, pre-HE, combined HE, and IAC populations in 10.5 dpc Runx1+/+ and Runx1+/− littermates. P values indicate significant differences in the distributions of cells in pre-HE and HE in Runx1+/+ vs Runx1+/− samples based on proportion test. ***P < .001. (F) UMAP of E+HE+IAC cells from 10.5 dpc control embryos (cR1/+) and littermates ectopically expressing RUNX1 in all endothelial cells from the Rosa26 locus (Cre;cR1/+).11 Bars at bottom as in panel E. ***P < .001. (G) Limiting dilution assay to determine the frequency of HE in the CD44+ fraction of E+HE+IAC cells isolated from 10.5 dpc embryos (see supplemental Figure 2G for fluorescence-activated cell sorting plots). Shown are frequencies of cells that yielded hematopoietic cells (B220+, CD19+, Mac1+, Gr1+, and/or CD41 and CD45) ex vivo. Frequencies were calculated by ELDA.64 Data represent 3 independent cell purifications and limiting dilution assays (mean ± standard deviation [SD], unpaired 2-tailed Student t test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/7/10.1182_blood.2020004801/3/m_bloodbld2020004801f3.png?Expires=1765884920&Signature=BdbI5EZ5OEe7Vj-fpbap8fnRGeQLZw5t2lD0SOKTv61YqrdvzdfJIw8mmTOg3FddI8k4iUvw8Ubm0BLbqnz2JrFzCgcUqgI8eHuoP064IWv2d8R2GWfN9lKhVW6pkT2QQK6Bfyq-dYhlyWa~RreaKS4Pm~0kJu-5qkC~ufnWZhFTE9TTQ3Gr5N9fCgGIZ~RleF7WRbTxqIrryddGL8E3hkvwS67kI7TKEjZncKAOcoIrTOnQvaPys4TQmicKCMQNHi3YxaAxAePzUj0d5dZ3GlvUo3dN13L1OD2bZt-ufYrVkKWp0vhVXLonSJT-pL~yypc4Sc4E~fLMrfyhZkcl2Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Developmental bottleneck between pre-HE and HE cells. (A) UMAP of E10.5 E+HE+IAC cells showing 9 cell types from Figure 2A. (B) Expression of key markers of clusters, including Hey2 in conflux AE and pre-HE, Cd44 in conflux AE, pre-HE, HE, and IACs; Ptprc in IACs; Gfi1 and Runx1 in HE and IACs; and high levels of Sox17 in conflux AE and pre-HE, with downregulation in HE. Note Runx1 is expressed at low levels in all subsets of endothelial cells. (C) Velocyto analysis revealing different differentiation dynamics along the EHT in ED10.5 E+HE+IAC cells. To the right is an enlargement velocity of pre-HE cells that have accumulated at the bottleneck between pre-HE and HE. (D) Activity of pathways from Kyoto Encyclopedia of Genes and Genomes database, computed for each cell using the AUCell method.63 (E) UMAP of E+HE+IAC cells from 10.5 dpc Runx1+/+ and Runx1+/− littermates. Bars at bottom depict the distribution of cells between conflux AE, pre-HE, combined HE, and IAC populations in 10.5 dpc Runx1+/+ and Runx1+/− littermates. P values indicate significant differences in the distributions of cells in pre-HE and HE in Runx1+/+ vs Runx1+/− samples based on proportion test. ***P < .001. (F) UMAP of E+HE+IAC cells from 10.5 dpc control embryos (cR1/+) and littermates ectopically expressing RUNX1 in all endothelial cells from the Rosa26 locus (Cre;cR1/+).11 Bars at bottom as in panel E. ***P < .001. (G) Limiting dilution assay to determine the frequency of HE in the CD44+ fraction of E+HE+IAC cells isolated from 10.5 dpc embryos (see supplemental Figure 2G for fluorescence-activated cell sorting plots). Shown are frequencies of cells that yielded hematopoietic cells (B220+, CD19+, Mac1+, Gr1+, and/or CD41 and CD45) ex vivo. Frequencies were calculated by ELDA.64 Data represent 3 independent cell purifications and limiting dilution assays (mean ± standard deviation [SD], unpaired 2-tailed Student t test).
Developmental bottleneck between pre-HE and HE cells. (A) UMAP of E10.5 E+HE+IAC cells showing 9 cell types from Figure 2A. (B) Expression of key markers of clusters, including Hey2 in conflux AE and pre-HE, Cd44 in conflux AE, pre-HE, HE, and IACs; Ptprc in IACs; Gfi1 and Runx1 in HE and IACs; and high levels of Sox17 in conflux AE and pre-HE, with downregulation in HE. Note Runx1 is expressed at low levels in all subsets of endothelial cells. (C) Velocyto analysis revealing different differentiation dynamics along the EHT in ED10.5 E+HE+IAC cells. To the right is an enlargement velocity of pre-HE cells that have accumulated at the bottleneck between pre-HE and HE. (D) Activity of pathways from Kyoto Encyclopedia of Genes and Genomes database, computed for each cell using the AUCell method.63 (E) UMAP of E+HE+IAC cells from 10.5 dpc Runx1+/+ and Runx1+/− littermates. Bars at bottom depict the distribution of cells between conflux AE, pre-HE, combined HE, and IAC populations in 10.5 dpc Runx1+/+ and Runx1+/− littermates. P values indicate significant differences in the distributions of cells in pre-HE and HE in Runx1+/+ vs Runx1+/− samples based on proportion test. ***P < .001. (F) UMAP of E+HE+IAC cells from 10.5 dpc control embryos (cR1/+) and littermates ectopically expressing RUNX1 in all endothelial cells from the Rosa26 locus (Cre;cR1/+).11 Bars at bottom as in panel E. ***P < .001. (G) Limiting dilution assay to determine the frequency of HE in the CD44+ fraction of E+HE+IAC cells isolated from 10.5 dpc embryos (see supplemental Figure 2G for fluorescence-activated cell sorting plots). Shown are frequencies of cells that yielded hematopoietic cells (B220+, CD19+, Mac1+, Gr1+, and/or CD41 and CD45) ex vivo. Frequencies were calculated by ELDA.64 Data represent 3 independent cell purifications and limiting dilution assays (mean ± standard deviation [SD], unpaired 2-tailed Student t test).
Runx1 expression is upregulated in ∼7% of pre-HE cells suggesting that RUNX1 levels regulate passage through the bottleneck (Figure 3B; supplemental Figure 6B). We tested this hypothesis using several approaches. First, we compared the distribution of cells between conflux AE, pre-HE, HE, and IAC in Runx1+/− and Runx1+/+ littermates by scRNA-Seq. We observed a 68% reduction in the proportion of HE and IAC cells in 10.5 dpc Runx1+/− compared with Runx1+/+ embryos, and a commensurate 56% increase in pre-HE, consistent with the hypothesis that RUNX1 levels regulate transit through the bottleneck (Figure 3E). We also performed the reciprocal experiment; ectopically expressing RUNX1 in all E cells by activating a conditional Runx1 complementary DNA in the Rosa26 locus (cR1) using an E-specific tamoxifen-inducible Cre driven from the vascular E cadherin (Cdh5) regulatory sequences (Cre).11 We previously showed that ectopic expression of RUNX1 in all E cells in Cre;cR1/+ embryos increased the frequency of functional HE cells compared with control embryos (cR1/+).11 scRNA-Seq analysis demonstrates this results from an increase in the proportion of HE cells and a proportionate decrease in pre-HE cells (Figure 3F), confirming that RUNX1 levels regulate the number of pre-HE cells that transit through the bottleneck to become HE. Second, we determined whether RUNX1 haploinsufficiency reduced the number of phenotypic HE cells by confocal microscopy. SOX17 is expressed in AE cells and promotes HE specification, whereas HE cells are RUNX1+SOX17low/−.30,31 The ratio of RUNX1+SOX17low/− HE cells vs RUNX1−SOX17+ AE cells in the dorsal aorta was significantly lower in 9.5 dpc Runx1+/− embryos compared with Runx1+/+embryos (supplemental Figure 8). Finally, we measured the frequency of functional HE cells within a purified population of CD44+ E+HE+IAC cells (supplemental Figure 2G), which are enriched for conflux AE, pre-HE, HE, and IAC32 (Figure 3B). RUNX1 haploinsufficiency reduced the frequency of functional HE cells by 77% (Figure 3G), consistent with the observed reduction in the scRNA-Seq experiment (Figure 3E). Together, these data confirm that RUNX1 levels regulate the number of pre-HE cells that transit through the bottleneck to become HE cells.
scATAC-Seq identifies putative Runx1 enhancers and transcription factor motifs that gain accessibility in pre-HE
To identify signals that may activate Runx1 expression in pre-HE, we performed paired scRNA-Seq and scATAC-Seq on 10.5 dpc CD44+ E+HE+IAC cells to identify Runx1 enhancers and the stages they are accessible. High-quality open chromatin profiles were obtained for 1670 cells, covering various cell types from E to IAC (supplemental Figure 9). The joint embedding of scRNA-Seq and scATAC introduced a gap between pre-HE and IAC cells on the UMAP. This results from the developmental bottleneck around ED10.5 that causes an underrepresentation of HE cells connecting pre-HE and IAC in some samples (Figure 4A). We devised a computational approach that matches scATAC-Seq clusters with scRNA-Seq clusters (Figure 4A; supplemental Figure 10), and subsequently linked enhancers with their target promoters (Figure 4B). Accuracy of our method was benchmarked using known hematopoietic and endothelial enhancers (Figure 4C-D). We applied chromVar33 to assess differential transcription factor (TF) binding patterns along the EHT trajectory (Figure 4E). Results show strong correlation with the TF expression patterns and are consistent with pathway analyses from scRNA-Seq data. For example, strong TCF/LEF binding activity was detected in Wnthi E that abruptly decreased in conflux AE (Figure 4E-F). SOX and FOX binding sites are mostly open in conflux AE and pre-HE. Binding sites for a large group of TFs had increased accessibility beginning at the pre-HE stage, including HES1, GATA, SMAD, and TFs such as MECOM, EGR1, and YY1 that regulate HSC homeostasis,34-36 with the latter group suggesting that an HSC-specific transcriptional program may initiate at the pre-HE stage.
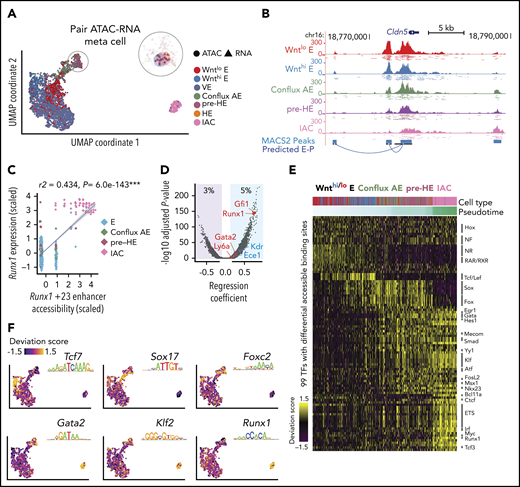
Joint scRNA-Seq and scATAC-Seq analysis of bottleneck populations. (A) UMAP of 1637 cells from scRNA-Seq and 1,186 cells from scATAC-Seq, aligned using Seurat algorithm with a custom defined gene-by-cell activity score matrix (see supplemental Methods). The number of HE cells was too few to be resolved by UMAP and was clustered with pre-HE. To gain enough statistical power for predicting E-P, we pooled reads from 10 nearest neighbors as “meta cells,” and paired scATAC meta cells to nearby scRNA meta cells. Additional details can be found in the supplemental Methods section. (B) UCSC genome browser tracks showing open chromatin signal of Cldn5 promoter and its predicted enhancers. Dots below each aggregated signal track represent signal from 50 sampled cells of each type. (C) Linear regression shows high correlation between Runx1 +23 enhancer chromatin accessibility and Runx1 expression levels (z-score transformed). Each point represents a paired ATAC-RNA meta cell in panel A, with pooled RNA expression on the y-axis and pooled enhancer accessibility on the x-axis. (D) Prediction of E-P interaction using linear regression. Predictions (points in blue shaded area, 5% of total candidate interactions) were made using P < .01 and regression coefficient >0.1. We recapitulated the majority of known E-P interactions that function during EHT, with the Runx1 +23 enhancer65 and Gfi1 enhancer66 among the top predictions. (E) TF binding patterns among called scATAC-Seq peaks assessed using chromVar,33 which defines a deviation score reflecting the accessibility change at binding sites of each TF across all cells. Binding sites were determined using DNA motif scan on the called enhancers, which does not discriminate TFs in the same family with very similar motifs. Top significant TFs based on Mann-Whitney U test are plotted for each stage. (F) ChromVar deviation score for selected TF motifs plotted on the UMAP, showing specific binding pattern for Tcf7 in Wnthi E, Sox17 in conflux E, Foxc2 in pre-HE, Gata2, and Klf2 in both pre-HE and IAC. Runx1 binding sites are highly accessible after bottleneck, but also exhibit medium to high level of chromatin accessibility in some early stage cells.
Joint scRNA-Seq and scATAC-Seq analysis of bottleneck populations. (A) UMAP of 1637 cells from scRNA-Seq and 1,186 cells from scATAC-Seq, aligned using Seurat algorithm with a custom defined gene-by-cell activity score matrix (see supplemental Methods). The number of HE cells was too few to be resolved by UMAP and was clustered with pre-HE. To gain enough statistical power for predicting E-P, we pooled reads from 10 nearest neighbors as “meta cells,” and paired scATAC meta cells to nearby scRNA meta cells. Additional details can be found in the supplemental Methods section. (B) UCSC genome browser tracks showing open chromatin signal of Cldn5 promoter and its predicted enhancers. Dots below each aggregated signal track represent signal from 50 sampled cells of each type. (C) Linear regression shows high correlation between Runx1 +23 enhancer chromatin accessibility and Runx1 expression levels (z-score transformed). Each point represents a paired ATAC-RNA meta cell in panel A, with pooled RNA expression on the y-axis and pooled enhancer accessibility on the x-axis. (D) Prediction of E-P interaction using linear regression. Predictions (points in blue shaded area, 5% of total candidate interactions) were made using P < .01 and regression coefficient >0.1. We recapitulated the majority of known E-P interactions that function during EHT, with the Runx1 +23 enhancer65 and Gfi1 enhancer66 among the top predictions. (E) TF binding patterns among called scATAC-Seq peaks assessed using chromVar,33 which defines a deviation score reflecting the accessibility change at binding sites of each TF across all cells. Binding sites were determined using DNA motif scan on the called enhancers, which does not discriminate TFs in the same family with very similar motifs. Top significant TFs based on Mann-Whitney U test are plotted for each stage. (F) ChromVar deviation score for selected TF motifs plotted on the UMAP, showing specific binding pattern for Tcf7 in Wnthi E, Sox17 in conflux E, Foxc2 in pre-HE, Gata2, and Klf2 in both pre-HE and IAC. Runx1 binding sites are highly accessible after bottleneck, but also exhibit medium to high level of chromatin accessibility in some early stage cells.
Runx1 contains 2 promoters, an upstream P1 promoter that is first used in committed HSPCs, and a more proximal P2 promoter that is active in HE and HSPCs.37 Consistent with this, P1 first becomes accessible in IAC cells, whereas P2 is accessible in all endothelial cells including pre-HE (Figure 5A), which may permit or contribute to the stochastic Runx1 expression observed in a subset of E cells (Figure 3B). Using our computational approach, we predicted 27 enhancer-promoter (E-P) interactions, which recapitulate 11 of 22 previously identified E-Ps based on chromosome conformation capture assays38,39 (Figure 5A; supplemental Methods). All of the predicted enhancers exhibit higher coaccessibility with P1 compared with P2; therefore, only E-Ps to P1 are indicated. A significance plot of the predicted E-Ps reveals several enhancers whose chromatin openness is significantly correlated with Runx1 expression, including the Runx1 +23 enhancer (Figure 5B). Several of the predicted enhancers exhibit stage-specific coaccessibility with the P1 promoter (Figure 5C). Interestingly, 1 candidate enhancer located 371 kb upstream of Runx1 P1 was accessible only in pre-HE and IACs, and not in other E cell populations (Figure 5A,C). This candidate enhancer was previously shown by circular chromosome conformation capture sequencing to interact with the +23 enhancer and P1 in a hematopoietic progenitor cell line.38 The −371 enhancer drove expression of a reporter gene in the intermediate cell mass and posterior blood island of zebrafish embryos, both of which are sites of hematopoietic ontogeny.38 The scATAC-Seq signal encompassing the −371 enhancer begins to increase in conflux AE cells and reaches a maximum in pre-HE cells (Figure 5A,D). This change in accessibility in pre-HE coincides with the activation of Runx1 expression in a subset of pre-HE cells (Figure 5D). However, unlike the +23 enhancer, the chromatin accessibility of the −371 enhancer subsequently decreases in IAC cells and is no longer open in FL-HSCs (Figure 5A). The candidate −371 enhancer contains GATA, STAT, and JUN motifs, indicating that GATA2 and cytokine and/or inflammatory signaling may contribute to the opening of this enhancer in pre-HE (Figure 5A). An independent coexpression analysis based on the scRNA-Seq data reveals that these factors form a coexpression gene module that precedes and correlates with Runx1 expression (Figure 5E), suggesting they may cooperatively regulate Runx1 expression. Notably, neither the −371 nor the +23 enhancers contain SOX motifs, which are recognized by a repressor of Runx1 expression, Sox17.31 Other TF motifs enriched in the 27 called Runx1 enhancers include ETS, FOX, SOX, KLF/SP, RUNX, and SMAD, which are recognized by TFs with well-documented roles in HSPC formation.40-42
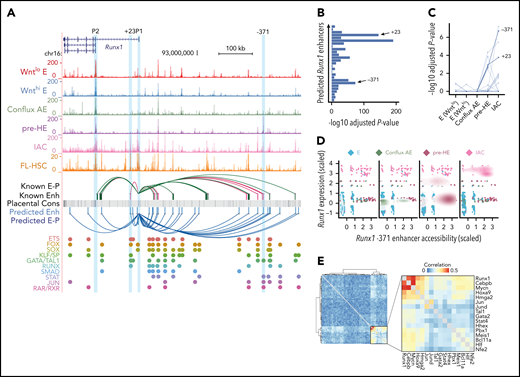
Developmental-stage-specific enhancers of Runx1. (A) UCSC genome browser tracks showing open chromatin signal for each of the populations. Tracks from E to IAC are cumulative scATAC-Seq signals (per-base unique fragment coverage) normalized by the number of cells in that population. Tracks for FL-HSC are bulk ATAC-Seq data from Chen et al.39 Experimentally validated enhancers and E-Ps from Marsman et al38 are shown in magenta. Enhancers and E-P links from Chen et al39 are shown in dark green. E-P links were inferred based on linear regression on paired scRNA-scATAC meta cells (supplemental Methods). Placental mammal conservation by PhastCons score is shown as a gray track. For each of the inferred enhancers, we scanned for known motifs from CIS-BP database and grouped TFs from the same family having similar motifs. Motif hits of several previously reported early hematopoietic TFs are highlighted below the track. (B) Distribution of linear regression P values for predicted Runx1 enhancers. Highly significant peaks include the validated +23 and −371 enhancers. The most significant peak is ∼3.6 kb downstream of P1. (C) Coaccessibility of Runx1 P1 promoter and its predicted linked enhancers in each cell type. P values for coaccessibility in each cell type were computed using Fisher exact test with multiple testing correction. (D) Stage-specific chromatin accessibility of Runx1 −371 enhancer and Runx1 expression levels (z-score transformed). Each point in the scatter plot represents a paired ATAC-RNA meta cell in Figure 5A, with pooled RNA expression on the y-axis and pooled enhancer accessibility on the x-axis. A 2-dimensional density plot is superimposed on the scatter plot. (E) Coexpression of transcription factors that have binding motifs at Runx1 enhancers and whose expression precedes Runx1. Correlations were computed using gene expression matrix including conflux E, pre-HE, and HE cells. TFs with Pearson correlation with Runx1 <0.05 were removed. Hierarchical clustering was performed on the correlation matrix and a strong TF coexpression module was highlighted.
Developmental-stage-specific enhancers of Runx1. (A) UCSC genome browser tracks showing open chromatin signal for each of the populations. Tracks from E to IAC are cumulative scATAC-Seq signals (per-base unique fragment coverage) normalized by the number of cells in that population. Tracks for FL-HSC are bulk ATAC-Seq data from Chen et al.39 Experimentally validated enhancers and E-Ps from Marsman et al38 are shown in magenta. Enhancers and E-P links from Chen et al39 are shown in dark green. E-P links were inferred based on linear regression on paired scRNA-scATAC meta cells (supplemental Methods). Placental mammal conservation by PhastCons score is shown as a gray track. For each of the inferred enhancers, we scanned for known motifs from CIS-BP database and grouped TFs from the same family having similar motifs. Motif hits of several previously reported early hematopoietic TFs are highlighted below the track. (B) Distribution of linear regression P values for predicted Runx1 enhancers. Highly significant peaks include the validated +23 and −371 enhancers. The most significant peak is ∼3.6 kb downstream of P1. (C) Coaccessibility of Runx1 P1 promoter and its predicted linked enhancers in each cell type. P values for coaccessibility in each cell type were computed using Fisher exact test with multiple testing correction. (D) Stage-specific chromatin accessibility of Runx1 −371 enhancer and Runx1 expression levels (z-score transformed). Each point in the scatter plot represents a paired ATAC-RNA meta cell in Figure 5A, with pooled RNA expression on the y-axis and pooled enhancer accessibility on the x-axis. A 2-dimensional density plot is superimposed on the scatter plot. (E) Coexpression of transcription factors that have binding motifs at Runx1 enhancers and whose expression precedes Runx1. Correlations were computed using gene expression matrix including conflux E, pre-HE, and HE cells. TFs with Pearson correlation with Runx1 <0.05 were removed. Hierarchical clustering was performed on the correlation matrix and a strong TF coexpression module was highlighted.
Two waves of HSPCs form in the IACs
We also examined the transition of HE to IAC cells and the composition of IAC cells. Principal component analysis (PCA) depicts a sharp U-turn as HE differentiates into IAC cells, reflective of a marked decrease in AE gene expression and activation of hematopoietic genes (Figure 6A). For example, the AE-specific gene Gja5 is primarily expressed on the HE side of the trajectory, whereas expression of Spn (encoding CD43), Ptprc (encoding CD45), and the Rho GTPase Rac2 rapidly increases in IAC cells (Figure 6B; supplemental Figure 11A-B). Transient expression of the chromatin remodeling protein Nupr1 occurs at the U-turn, whereas Hey1 and Sox17 transcripts significantly diminish as IAC cells mature (Figure 6B; supplemental Figure 11B).
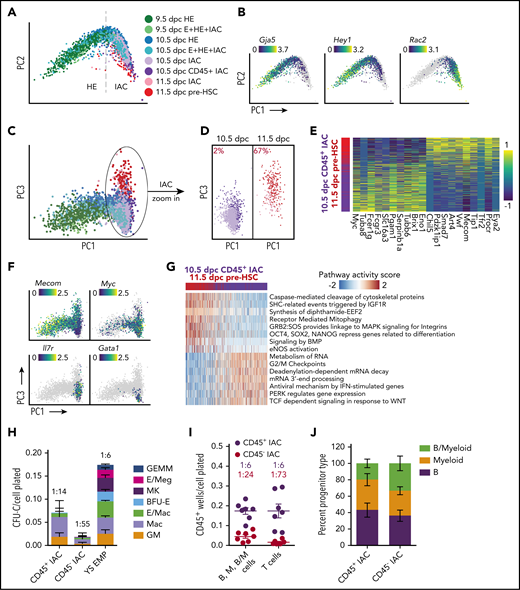
Two waves of CD45+ HSPCs in IAC cells. (A) PCA plot of a subset of data containing IACs, illustrating the trajectory of IAC differentiation from HE along the PC1 axis. (B) Expression of Gja5, Hey1, and Rac2 illustrating the maturation of IAC cells along the trajectory. (C) PCA plot showing the separation of 10.5 and 11.5 dpc IAC cells along the PC3 axis. (D) 10.5 and 11.5 dpc IAC cells, 10.5 dpc CD45+ IAC cells, and 11.5 dpc pre-HSCs plotted separately to visualize their relative distribution along the PC3 axis. A k-nearest-neighbor classifier (k = 3 with PC1-10 as feature input) was trained using 10.5 dpc CD45+ IAC cells and 11.5 dpc pre-HSCs to determine the fraction of pre-HSCs (red) in 10.5 and 11.5 dpc IAC cells. (E) Heatmap showing top differentially expressed genes in 10.5 dpc CD45+ IAC cells vs 11.5 dpc pre-HSCs. (F) Preferential expression of Mecom in 11.5 dpc pre-HSCs and IAC cells vs Myc, Il7r, and Gata1 in 10.5 dpc CD45+ IAC enriched for lymphomyeloid-biased progenitors and in 10.5 dpc IAC cells. (G) Reactome pathway analysis comparing 11.5 dpc pre-HSC and 10.5 dpc CD45+ IAC cells. Color indicates pathway activity score computed using the AUCell package.63 (H) Methylcellulose (colony forming unit-culture, CFU-C) assay performed in the presence of stem cell factor (SCF), interleukin 3 (IL-3), IL-6, and erythropoietin (EPO) to measure the frequency of committed erythroid and myeloid progenitors in 10.5 dpc CD45+ IAC cells, CD45− IAC cells, and 9.5 dpc yolk sac EMPs. BFU-E, burst forming unit-erythroid; GEMM, granulocyte/erythroid/monocyte/megakaryocyte; GM, granulocyte/macrophage; Mac, macrophage; MK, megakaryocyte. Values and error bars are mean ± SD; n= 3 experiments. Frequencies of total progenitors are indicated above the bars. (I) Limiting dilution assays on OP9 stromal cells to determine the frequencies of progenitors in purified 10.5 dpc CD45+ IAC and 10.5 dpc CD45− IAC cells yielding B (CD45+CD19+B220mid/lo), myeloid (M) (Gr1+Mac1+ or Gr1+Mac1−), and B+myeloid (B/M) cells in culture. Also shown are frequencies of progenitors in purified 10.5 dpc CD45+ IAC and 10.5 dpc CD45− IAC cells that produced T cells (CD90+ CD25+) when cultured on OP9 cells expressing the Notch ligand delta-like 1. Error bars; mean ± SD. Values and error bars are mean ± SD; n = 7. Frequencies of all progenitors are indicated above the bars. (J) Percentage of wells at the limiting cell dose containing B, M, or B/M cells from experiments in panel I. Values and error bars are mean ± SD; n = 8 experiments.
Two waves of CD45+ HSPCs in IAC cells. (A) PCA plot of a subset of data containing IACs, illustrating the trajectory of IAC differentiation from HE along the PC1 axis. (B) Expression of Gja5, Hey1, and Rac2 illustrating the maturation of IAC cells along the trajectory. (C) PCA plot showing the separation of 10.5 and 11.5 dpc IAC cells along the PC3 axis. (D) 10.5 and 11.5 dpc IAC cells, 10.5 dpc CD45+ IAC cells, and 11.5 dpc pre-HSCs plotted separately to visualize their relative distribution along the PC3 axis. A k-nearest-neighbor classifier (k = 3 with PC1-10 as feature input) was trained using 10.5 dpc CD45+ IAC cells and 11.5 dpc pre-HSCs to determine the fraction of pre-HSCs (red) in 10.5 and 11.5 dpc IAC cells. (E) Heatmap showing top differentially expressed genes in 10.5 dpc CD45+ IAC cells vs 11.5 dpc pre-HSCs. (F) Preferential expression of Mecom in 11.5 dpc pre-HSCs and IAC cells vs Myc, Il7r, and Gata1 in 10.5 dpc CD45+ IAC enriched for lymphomyeloid-biased progenitors and in 10.5 dpc IAC cells. (G) Reactome pathway analysis comparing 11.5 dpc pre-HSC and 10.5 dpc CD45+ IAC cells. Color indicates pathway activity score computed using the AUCell package.63 (H) Methylcellulose (colony forming unit-culture, CFU-C) assay performed in the presence of stem cell factor (SCF), interleukin 3 (IL-3), IL-6, and erythropoietin (EPO) to measure the frequency of committed erythroid and myeloid progenitors in 10.5 dpc CD45+ IAC cells, CD45− IAC cells, and 9.5 dpc yolk sac EMPs. BFU-E, burst forming unit-erythroid; GEMM, granulocyte/erythroid/monocyte/megakaryocyte; GM, granulocyte/macrophage; Mac, macrophage; MK, megakaryocyte. Values and error bars are mean ± SD; n= 3 experiments. Frequencies of total progenitors are indicated above the bars. (I) Limiting dilution assays on OP9 stromal cells to determine the frequencies of progenitors in purified 10.5 dpc CD45+ IAC and 10.5 dpc CD45− IAC cells yielding B (CD45+CD19+B220mid/lo), myeloid (M) (Gr1+Mac1+ or Gr1+Mac1−), and B+myeloid (B/M) cells in culture. Also shown are frequencies of progenitors in purified 10.5 dpc CD45+ IAC and 10.5 dpc CD45− IAC cells that produced T cells (CD90+ CD25+) when cultured on OP9 cells expressing the Notch ligand delta-like 1. Error bars; mean ± SD. Values and error bars are mean ± SD; n = 7. Frequencies of all progenitors are indicated above the bars. (J) Percentage of wells at the limiting cell dose containing B, M, or B/M cells from experiments in panel I. Values and error bars are mean ± SD; n = 8 experiments.
IACs contain pre-HSCs that cannot engraft adult mice directly, but can mature in vivo or ex vivo into adult-repopulating HSCs.43-45 Pre-HSCs are classified as type I or II based on CD45 expression: type I are CD45− and the more mature type II HSCs are CD45+.43 IACs 10.5 dpc contain only type I pre-HSCs, whereas 11.5 dpc IACs contain both type I and II pre-HSCs.43 Additionally, multiple progenitors with lymphoid, myeloid, lymphomyeloid, or multilineage potential emerge before or contemporaneously with pre-HSCs,1 at least a subset of which are CD45+.5,46,47 We compared 10.5 dpc CD45+ IAC cells that contain HSC-independent progenitors and lack pre-HSCs to 11.5 dpc CD45+CD27+CD144+ IAC cells enriched for type II pre-HSCs (11.5 dpc pre-HSCs)43,48 to determine their developmental relationship (supplemental Figure 2C-D). The 2 populations bifurcate in the third principal component of PCA plots; specifically, the majority of 10.5 dpc CD45+ IAC cells occupy one end of the PC3 axis, and 11.5 dpc pre-HSCs reside on the other end (Figure 6C-D). Pre-HSCs 11.5 dpc demonstrated a high correspondence with previously published data6 (supplemental Figure 12A). We determined the fraction of pre-HSCs in 10.5 and 11.5 dpc IAC cells using a k-nearest-neighbor classifier. About 2% of 10.5 dpc IAC cells were found to be molecularly similar to 11.5 dpc type II pre-HSCs; this fraction of pre-HSCs increases to 67% in 11.5 dpc IAC cells (Figure 6D), consistent with previous limiting dilution assay results demonstrating an increase in functional pre-HSCs between 10.5 and 11.5 dpc.45 To determine the fate bias of cells from earlier stages, we used Palantir and FateID.49,50 T-SNE plot generated by Palantir trajectory analysis shows a bifurcation pattern similar to the PCA result (supplemental Figure 11C). Distribution of the fate probabilities suggests that compared with 9.5 dpc HE, 10.5 dpc HE has higher probability of choosing pre-HSC fate, and 11.5 dpc IACs contain more pre-HSC-like cells than 10.5 dpc IACs (1-sided Kolmogorov-Smirnov test; supplemental Figure 11D-E).
The 974 genes more highly expressed in 11.5 dpc pre-HSCs compared with 10.5 dpc CD45+ IAC cells include known markers of pre-HSCs and/or HSCs (Eya2, Procr, Cd27, and Mecom),6,34,48,51,52 whereas the 877 genes upregulated in 10.5 dpc CD45+ IAC cells include proliferation related genes (Myc) and lymphomyeloid associated genes (Il7r, Fcer1g) (Figure 6E-F; supplemental Table 5). Among the differentially expressed genes, some transcription factors, such as Myc, Klf2, Smad7, Mecom, Meis2, and Nfix, are expressed in HE cells, and show strong bifurcation in expression as cells become IAC cells (supplemental Figure 11F). Pathway analysis suggests 11.5 dpc pre-HSCs gain stem cell-specific features such as "OCT4, SOX2, NANOG represses genes related to differentiation," whereas pathways associated with 10.5 dpc CD45+ IAC cells are associated with cell cycle and/or related to a specific hematopoietic lineage, such as “TCF dependent signaling in response to WNT” (Figure 6G; supplemental Table 5). Interestingly, 11.5 dpc pre-HSCs, although sampled 1 day later in development compared with 10.5 dpc CD45+ IAC cells, retain many pathways from E/HE stages, such as “Signaling by BMP” and “eNOS activation”, suggesting a relatively slow shutdown of the E/HE program in pre-HSCs. In contrast, subsets of 10.5 dpc CD45+ IAC cells show lineage-specific differentiation bias; Il7r is upregulated in 26% and Gata1 in 3% of 10.5 dpc CD45+ IAC cells (Figure 6F).
Previous scRNA-Seq studies identified committed progenitors in 10.5 and 11.5 dpc IACs but concluded they were contaminating EMPs, likely originating from the yolk sac, that had been circulating in the blood and became attached to the IACs.5,6 We addressed the possibility that the 10.5 dpc CD45+ IAC cells we profiled are contaminating YS EMPs. Direct comparison shows 10.5 dpc CD45+ IAC cells and 9.5 dpc YS-EMP are molecularly and functionally distinct (Figure 6H; supplemental Figure 12C). CD45+ 10.5 dpc IAC cells contained progenitors of macrophages and granulocytes/monocytes, but very few erythroid or megakaryocytic progenitors compared with 9.5 dpc YS-EMPs (Figure 6H). CD45+ 10.5 dpc IACs have potent lymphoid potential; limiting dilution assays revealed a high frequency of cells (1:6) capable of producing B cells following culture on OP9 stromal cells or T cells on OP9 expressing the Notch ligand delta-like 1 (Figure 6I-J). CD45− 10.5 dpc IAC cells also contained progenitors with lymphoid and myeloid potential, although their frequency was lower than in the 10.5 dpc CD45+ IAC population. In summary, 10.5 dpc CD45+ IAC cells represent a distinct wave of lymphomyeloid-biased progenitors in IACs that appear before 11.5 dpc type II pre-HSCs.
Discussion
Our single-cell analyses provide new insights into the process by which endothelial cells differentiate into pre-HSCs. First, we define a precursor of HE we have named pre-HE, in which multiple molecules and pathways known to regulate HSPC formation appear to act. Also, through trajectory analyses and genetic perturbation experiments, we identified a bottleneck separating pre-HE from HE, indicative of a developmental barrier that must be overcome at that transition. It is long known that embryonic hematopoiesis is exquisitely sensitive to Runx1 dosage, as reduced Runx1 dosage decreases the number of HE cells, IACs, and committed hematopoietic progenitors in the embryo.12,53,54 Our scRNA-Seq analyses show that the deficits caused by reduced Runx1 dosage are caused, at least in part, by the inefficient transition of pre-HE to HE cells. The molecular underpinnings of the bottleneck at the pre-HE to HE transition are not known. One possibility is that Runx1 expression may be actively repressed in the majority of pre-HE cells by TFs such as Sox17,31,55 which is highly expressed in pre-HE, and binding sites that are accessible in pre-HE. A requirement for chromatin remodeling may also be a limiting factor in pre-HE because multiple epigenetic regulatory proteins have been shown to affect Runx1 expression in HE, some of which may act at the pre-HE to HE transition.56
Before HE, Runx1 is expressed at low levels in a subset of endothelial cells, consistent with the chromatin accessibility of the P2 promoter and of several Runx1 enhancers in endothelial cells. Runx1 expression in endothelial cells appears to be stochastic; it then becomes elevated in a subset of pre-HE cells, and is uniformly high in HE and IACs. The mechanism by which Runx1 expression is activated in a subset of pre-HE cells is not known, but our experiments provide some clues. scATAC-Seq revealed that a distal enhancer in Runx1(−371), previously validated in zebrafish transgenic embryos and conserved in mammals,38 first becomes accessible in pre-HE. Highly conserved TF motifs in the −371 enhancer include GATA, STAT, and JUN, implying that TFs that bind these motifs may play a role in opening the enhancer in pre-HE. Gata2 expression is activated in a pulsatile manner in endothelial cells in the DA,57 which may contribute to the stochastic expression of Runx1 in arterial endothelial cells. STAT and JUN motifs are recognized by TFs that are effectors of inflammatory signaling pathways, including type I and II interferons, and tumor necrosis factor, all of which promote HSPC formation from arterial endothelium.28,58,59 Hence well-known signaling pathways known to promote later Runx1 expression in HE could potentially initiate Runx1 expression in a subset of pre-HE cells by activating the candidate −371 pre-HE enhancer. At later stages, in IACs and FL-HSCs, multiple additional enhancers, including the +23 enhancer gain accessibility and interact with the P1 promoter to further elevate Runx1 expression.
A second important concept gleaned from our data are that the IACs contain at least 2 distinct HSPC subtypes, committed lymphomyeloid-biased progenitors and pre-HSCs, which can be distinguished molecularly. These appear sequentially, with CD45+ lymphomyeloid-biased progenitors preceding the formation of type II pre-HSCs. The mechanisms underlying the generation of these 2 types of HSPCs is of great interest. It is not known, for example, if they independently differentiate from an equivalent population of immature IAC cells. Alternatively, they may be derived from distinct populations of HE cells. Our cell fate probability analysis suggests that 10.5 dpc HE is more likely to assume pre-HSC fate than 9.5 dpc HE, which is consistent with the observation that 11.5 dpc IACs contain more pre-HSCs than 10.5 dpc IACs. The bifurcation of fate may be partially driven by early differential expression of transcription factors specific to pre-HSCs or lymphomyeloid-biased progenitors. The lymphomyeloid-biased progenitors are more developmentally “mature” compared with the type II pre-HSCs, suggesting that they are more driven toward terminal differentiation. A similar population of lymphomyeloid-restricted progenitors that originates in the yolk sac colonizes the FL and thymus before HSCs.60,61 Lymphomyeloid-biased progenitors in the arterial IACs may serve a similar function.
The earlier emergence of lymphomyeloid-biased progenitors in the arteries may have implications for ongoing efforts to generate pre-HSCs from ES cells. The acquisition of lymphoid potential is often used as a surrogate for pre-HSC formation. However, it is possible that conditions favoring the production of this earlier population of committed lymphomyeloid progenitors may be suboptimal for the later formation of pre-HSCs. If this is the case, then inhibiting the differentiation of lymphomyeloid progenitors in ES cell cultures may improve pre-HSC production ex vivo.
Data and analysis from this study can be interactively explored using a single-cell EHT data explorer based on VisCello13 and R shiny62 (https://github.com/qinzhu/VisCello.eht).
Data generated during this study have been deposited in the Gene Expression Omnibus with the accession number GSE137117. The endothelial to hematopoietic cell transition (EHT) atlas can be interactively explored using VisCello 13 (https://github.com/qinzhu/VisCello.eht).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The Runx1:GFP mice were kindly provided by James Downing. The authors thank Sumedha Bagga for editorial assistance, and Youtao Lu and Junhyong Kim for suggestions on analyses.
This work was supported by grants from the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (R01HL091724) (N.A.S.), NIH National Institute of Allergy and Infectious Diseases (R21AI133261) (N.A.S.), NIH National Human Genome Research Institute (R01HG006130 and R01 GM108716) (K.T.), and the Pennsylvania Department of Health (K.T. and N.A.S.). The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions. L.B. is supported by a grant from the NIH National Heart, Lung, and Blood Institute (T32HL007439), M.M. by NIH National Institute of Diabetes and Digestive and Kidney Diseases (T32DK007780), and E.D.H. by NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development (T32HD083185).
Authorship
Contribution: Q.Z. conducted the bioinformatics analyses and wrote the manuscript; P.G. and C.C. performed the scRNA-Seq and scATAC-Seq experiments; J.T., L.B., Y.L., M.M., and E.D.H. purified and functionally validated the cell populations; Y.U., W.Y., and B.H. conducted additional bioinformatics analyses; and N.A.S. and K.T. directed the project and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for Y.L. is Department of Veterinary Medicine and Institute of Preventive Veterinary Sciences, Zhejiang University, Hangzhou, Zhejiang, China.
The current affiliation for B.H. is Department of Pediatrics, College of Medicine, Pennsylvania State University, Hershey, PA.
Correspondence: Kai Tan, Division of Oncology and Center for Childhood Cancer Research, 3501 Civic Center Blvd, Children’s Hospital of Philadelphia, Philadelphia, PA 19104; e-mail: tank1@e-mail.chop.edu; and Nancy A. Speck, Abramson Family Cancer Research Institute and Department of Cell and Developmental Biology, 421 Curie Blvd, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; e-mail: nancyas@upenn.edu.
