

In this issue of Blood, demonstrate that in β-thalassemia hematopoietic stem cells (HSCs) are impaired due to defective interaction with the bone marrow microenvironment (BME), a defect that can be rescued by administration of parathyroid hormone (PTH).1
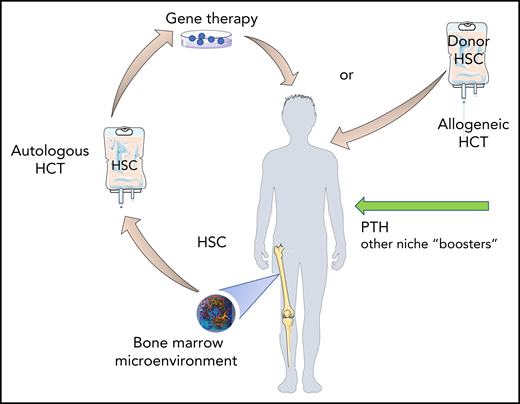
Protecting the seed, fertilizing the soil. Administration of PTH, or other “niche” boosters, may restore altered BM homeostasis in β-thalassemia patients resulting in more “fitted” HSC for gene therapy and a more permissive BM niche for engraftment of donor cells. Design elements were obtained from Smart Servier Medical Art (https://smart.servier.com/) and modified as needed. The bone marrow microenvironment snapshot is an image generated by the author.
Protecting the seed, fertilizing the soil. Administration of PTH, or other “niche” boosters, may restore altered BM homeostasis in β-thalassemia patients resulting in more “fitted” HSC for gene therapy and a more permissive BM niche for engraftment of donor cells. Design elements were obtained from Smart Servier Medical Art (https://smart.servier.com/) and modified as needed. The bone marrow microenvironment snapshot is an image generated by the author.
This study illuminates the fact that β-thalassemia not only impairs red cell production but also affects global bone marrow (BM) homeostasis, including HSCs and their BM niche, a concept likely relevant to other hemoglobinopathies.
In the genetic disorder β-thalassemia, lack of β-globin synthesis results in peripheral hemolysis, anemia, and ineffective erythropoiesis. The severe form of this disease, β-thalassemia major, requires lifelong transfusions and iron chelation therapy. A major consequence of the disease and treatment is iron overload, which inflicts chronic oxidative organ damage over time, reducing life span.2 Sickle cell disease is another severe hemoglobinopathy resulting from defective β-globin production. It is characterized by anemia and vasoocclusive crises, which cause ischemic and oxidative organ damage and decrease life span.2 Despite differences in etiology, the sequelae of altered erythropoiesis in both diseases inflict chronic and systemic injury to all organs, including the BM.
For β-thalassemia major and severe sickle cell disease, hematopoietic cell transplantation (HCT) from an HLA-identical sibling donor is currently the only curative therapeutic option.3 However, few patients have potentially HLA-matched siblings; the incidence of graft failure in the haploidentical setting remains high, and the risk of graft-versus-host-disease is considerable in HLA-matched unrelated HCT.3 Gene therapy via engineering autologous hematopoietic stem/progenitor cells with corrective approaches has significant potential for a cure, and clinical safety and efficacy trials are ongoing in both diseases.4 However, significant challenges persist, including preserving the fitness of HSCs for gene modification in patients with severe disease, mobilizing enough HSCs, and maintaining engraftment in a BM niche exposed to recurrent injury. Although the BM is the organ targeted for a cure, little is currently known about how the cumulative effects of hemolysis, iron accumulation, and inflammatory signals caused by these hemoglobinopathies damage the BME and impair HSC functions.
In this elegant study, Aprile and colleagues are the first to test whether β-thalassemia affects HSC function. Using a mouse model of β-thalassemia that recapitulates features of β-thalassemia major in patients, the group made some important discoveries. They observed that β-thalassemic mice have decreased numbers of HSCs, which are characterized by decreased quiescence and increased stress responses. In key transplant experiments, the authors show that β-thalassemic HSCs exhibit also impaired functions, such as repopulating ability and stem cell self-renewal, when transplanted into β-thalassemic recipients. However, these defects are rescued when β-thalassemic HSCs are transplanted into control recipients. This suggests that HSC dysfunction in β-thalassemic mice is not intrinsic and is reversible, and that, although perpetuated by a β-thalassemic BME, it can be rescued by interaction with a healthy BME. The authors also show that β-thalassemic mice exhibit an altered BME, characterized by reduced bone mass, decreased osteoblast (OB) anabolism, and low levels of PTH, similar to observations made in β-thalassemic patients.2,5 Premised upon the known role of PTH and the OB niche in maintaining HSC function,6 the authors focus their studies on OBs and mesenchymal stem cells (MSCs) in the BM of β-thalassemic mice and demonstrate lower levels of osteopontin (OPN) and of the Notch ligand Jagged1 (J1), both known regulators of HSC quiescence and stemness.7 The authors propose a working model in which low expression of PTH in β-thalassemia impairs bone homeostasis, resulting in decreased expression of OPN and J1 by OBs and consequent loss of quiescence and impairment of Notch signaling in HSCs. As proof of concept, they administer PTH to β-thalassemic mice and demonstrate a remarkable normalization of bone density, which is associated with increased OPN and J1 expression in the BME and with restoration of HSC function.
A critical aspect of this work is that Aprile and colleagues validate their findings in patients’ samples and link HSC and bone alterations to PTH, the levels of which are lower in β-thalassemia patients compared with healthy individuals.2,5 They are the first to document molecular changes and cell-cycle alterations in HSCs from β-thalassemia patients, which they correlate with a BM niche marked by decreased OB function and low OPN and J1 expression. Although the analysis is conducted in a small number of patients, the data are consistent and pose a promising platform for future mechanistic studies of HSC function and their defective interaction with the BM niche in a larger pool of patients.
It is interesting that OPN and J1/Notch are the main molecules significantly altered in the authors’ analysis. Not surprisingly, these molecules are directly regulated by PTH in OBs.6 OPN is a robust promoter of HSC quiescence7 and Notch signaling regulates HSC responses during stress hematopoiesis.8 Although the role of Notch in HSC homeostasis is still controversial, this study opens the interesting possibility that J1/Notch interaction has an underestimated regulatory function in BM stress induced by hemoglobinopathies. However, it is also important to consider that the global impact of PTH on OB anabolism is likely to be most relevant in the reestablishment of BM homeostasis and rescue of HSCs, due to the collective contributions of other molecules provided by OBs to HSCs.6 Furthermore, previous work by this group identified defective MSCs in the BME of β-thalassemic patients due to iron overload,9 providing additional mechanisms accounting for overall BME dysfunction.
The marked positive effect of PTH on normalizing BM function in β-thalassemic mice, as seen in this study, holds the promise that PTH administration could help restore damaged BME/HSC interactions in β-thalassemic patients to improve current therapeutic strategies. This could include treatment with PTH before HSC harvest and before HCT to correct the BME, protect HSC function, and improve engraftment (see figure). After an initial enthusiasm for using PTH to stimulate HSCs and increase their engraftment upon transplantation, subsequent studies have been disappointing.10 However, the patients treated were a heterogeneous group; specific targeting of the β-thalassemic BM niche by PTH may be beneficial and successful, as these patients exhibit suboptimal PTH levels and altered bone homeostasis. A critical point will be to determine whether HSC dysfunction in humans is reversible as observed in the mouse model, given that years of HSC exposure to cumulative damage in the BME of patients may not be accurately reflected by weeks/months of HSC exposure to damaged BME in mice.
This work opens up new questions, and it is clear that a deeper understanding of HSC biology and the status of the BM niche and its components in these hemoglobinopathies is essential for the development of curative approaches, both for improving the success of allogeneic HCT and for effective translation of promising gene-correction strategies in autologous HCT. Aprile and colleagues bring new insights on the BME in β-thalassemia and advance the novel concept of targeting the BM niche to improve treatment outcomes in these diseases.
The journey has just begun.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal