

In this issue of Blood, 1 identified 2 distinct DNA methylation subtypes for Waldenström macroglobulinemia (WM) by performing methylation screens in 35 well-characterized patients using genome-wide BeadChip technology. In 24 patients, the data were supplemented by transcriptome and targeted DNA sequencing permitting a detailed, multi-omic characterization of WM. Although individual epigenetic events have been previously characterized, this is the first genome-wide characterization and represents an important milestone for understanding WM pathogenesis.
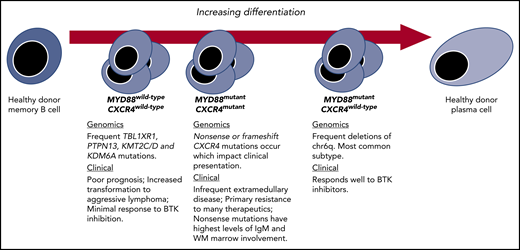
Genetic subtypes and the spectrum of differentiation in WM. Key genomic and clinical characteristics of WM disease based on revelations made possible by integrating transcriptome and epigenomic findings.
Genetic subtypes and the spectrum of differentiation in WM. Key genomic and clinical characteristics of WM disease based on revelations made possible by integrating transcriptome and epigenomic findings.
WM is an ideal disease model for a multi-omic approach as it features highly recurring somatic activating mutations, including MYD88 and CXCR4 mutations.2 Moreover, MYD88 and CXCR4 activating mutations are clinically relevant as they associate with important disease-presenting features and have prognostic and/or predictive treatment roles.3 A number of structural alterations have also been identified, the most common being the deletion of chromosome 6q (chr6q) in 30% to 50% of WM patients with mutated MYD88, but which are absent in those with wild-type MYD88.2,3 Deletions in chr6q are not unique to WM and are found in other lymphomas, and myeloma, and often occur with chromosome 6p gains. These highly recurring somatic events permit even a modest size multi-omic study such as the one reported by Roos-Weil et al to be informative.
Central to the findings of Roos-Weil et al was the finding that within mutated MYD88 WM patients, the methylome stratified patients into 2 camps: one with similar profiling to healthy donor (HD) memory B cells (MBC-like) and the other similar to HD plasma cells (PC-like). Those WM patients with MBC-like profiling showed DNA methylation changes that targeted functional domains related to transcriptional activation, whereas among those with PC-like profiling, broader losses in methylation that impacted repressed, heterochromatic, as well as intergenic regions were observed. The notion that certain WM patients have greater plasmacytic differentiation has been long recognized at the morphological and transcriptional level.4 However, the ability to so clearly define and characterize these groups by differences in methylation offers new and valuable insights, particularly the association of chr6q deletions with a PC-like clone. In previous work, we observed a lack of chr6q deletions in samples from WM patients with wild-type MYD88 by whole-genome and -exome sequencing and inferred that chr6q deletions represented a likely second hit following acquisition of mutated MYD88.2,5 Other studies speculated that chr6q deletions were critical to the transformation of immunoglobulin M (IgM) monoclonal gammopathy of undetermined significance (MGUS) to WM.6-8 The findings by Roos-Weil et al suggest that such clones, regardless of when and where chr6q deletions occurred, are destined for a PC-like clone.
CXCR4 mutations represent early somatic events that occur soon after the MYD88 mutation and are detectable even in those individuals with IgM MGUS.2 Previous transcriptional profiling of samples from patients with both MYD88 and CXCR4 mutations showed less plasmacytic differentiation compared with patients who were MYD88 mutated but wild-type for CXCR4.5 With the data now in hand from Roos-Weil et al, we can assert that CXCR4 mutations are characteristic of an earlier, MBC-like WM clone, whereas chr6q deletions are characteristic of a more differentiated PC-like clone. CXCR4 mutations have also been observed in MYD88 wild-type patients, which is notable as they represent the least differentiated form of WM.5,6 A number of clinically relevant disease features are associated with CXCR4 mutations, including lower rates of deep responses, and shorter progression-free survival with treatment using BTK inhibitors.3 Roos-Weil and colleagues provide an additional dimension challenging us to take into account the importance of the epigenome to such differences in therapeutic outcome.
The data presented by Roos-Weil et al also complement our previous work that examined methylation differences within WM based on mutation status using enhanced reduced representational bisulfite sequencing.9 As was observed in our RNASeq analysis, pairwise multidimensional scaling of the methylation status of the top 2000 high-variance promoters revealed segregation of samples from patients expressing MYD88mutant/CXCR4wild-type vs those with MYD88mutant/CXCR4mutant and MYD88wild-type/CXCR4wild-type. Promoter level analysis revealed 556 differentially methylated promoters in samples from MYD88mutant/CXCR4mutant patients of which 440 (78%) had increased methylation relative to MYD88mutant/CXCR4wild-type samples. These findings were consistent with greater hypomethylation associated with plasmacytic differentiation. Effected genes included IL15, GNAO1, HIF1A, SOCS6, PIK3R5, IRF8, and CD38. For MYD88wild-type/CXCR4wild-type samples, and 126 promoters showed significantly different methylation with only 28 (22%) demonstrating increased methylation relative to MYD88mutant/CXCR4wild-type samples. Effected promoters included NRIP1, FNBP1L, and PTK2 in these samples.
Although these observations may tempt thinking of WM as 2 separate diseases based on differentiation status, it is important to consider the crucial role that mutated MYD88 plays as the primary driver of prosurvival signaling. Indeed, when mutated MYD88 signaling was blocked, signaling through wild-type or mutated CXCR4 failed to rescue cells in the presence of CXCL12, the ligand for the CXCR4. Moreover, although chr6q deletions and CXCR4 mutations appear mutually exclusive, their gene expression profiles relative to MYD88 mutant WM patients lacking these events overlap significantly with a set of genes moving in the same direction with a similar log fold change.8 As both CXCR4 mutations and chr6q deletions are thought to be events occurring after the acquisition of MYD88, this shared signaling suggests a common path in the evolution to symptomatic WM, and possibly one that might be exploited therapeutically.
Although the findings by Roos-Weil et al provide great insights into the epigenome of WM, they also contribute to our understanding of the topography of the WM genome. The root cause of methylation differences within WM remains to be discerned and represents a critical research question relevant to not only WM pathogenesis but also therapeutic targeting. Transgenic modeling has revealed that methyltransferases such as DNMT3A/B impact B-cell activation, plasmacytic differentiation, and humoral immunity.10 Other contributors worthy of investigation include regulators of histone methylation such as KMT2A, KMT2D, and KDM6A that are frequently mutated in WM.2
In summary, Roos-Weil and colleagues have added a third dimension to our understanding of WM genomics, complementing revelations in the transcriptome and genome of WM, and allowing us to demarcate WM based on a spectrum of lymphoplasmacytic differentiation (see figure).
Conflict-of-interest disclosure: Z.R.H. has received honoraria from Janssen Pharmaceuticals. S.P.T. has received research funding and consulting fees from Pharmacyclics Inc, Janssen Pharmaceuticals, and Beigene.
