

Key Points
HyperD-ALL shows a delay in early mitosis at prometaphase associated with defects in chromosome alignment and segregation.
Impaired condensin complex leads to defective AURKB, triggering chromatid cohesion defects and mitotic slippage of HyperD-ALL blasts.

Abstract
B-cell acute lymphoblastic leukemia (ALL; B-ALL) is the most common pediatric cancer, and high hyperdiploidy (HyperD) identifies the most common subtype of pediatric B-ALL. Despite HyperD being an initiating oncogenic event affiliated with childhood B-ALL, the mitotic and chromosomal defects associated with HyperD B-ALL (HyperD-ALL) remain poorly characterized. Here, we have used 54 primary pediatric B-ALL samples to characterize the cellular-molecular mechanisms underlying the mitotic/chromosome defects predicated to be early pathogenic contributors in HyperD-ALL. We report that HyperD-ALL blasts are low proliferative and show a delay in early mitosis at prometaphase, associated with chromosome-alignment defects at the metaphase plate leading to robust chromosome-segregation defects and nonmodal karyotypes. Mechanistically, biochemical, functional, and mass-spectrometry assays revealed that condensin complex is impaired in HyperD-ALL cells, leading to chromosome hypocondensation, loss of centromere stiffness, and mislocalization of the chromosome passenger complex proteins Aurora B kinase (AURKB) and Survivin in early mitosis. HyperD-ALL cells show chromatid cohesion defects and an impaired spindle assembly checkpoint (SAC), thus undergoing mitotic slippage due to defective AURKB and impaired SAC activity, downstream of condensin complex defects. Chromosome structure/condensation defects and hyperdiploidy were reproduced in healthy CD34+ stem/progenitor cells upon inhibition of AURKB and/or SAC. Collectively, hyperdiploid B-ALL is associated with a defective condensin complex, AURKB, and SAC.
Introduction
B-cell acute lymphoblastic leukemia (B-ALL) is characterized by the accumulation of abnormal immature B-cell precursors (BCPs) in the bone marrow (BM), and is the most common pediatric cancer.1 B-ALL is a heterogeneous disease with distinct biological prognostic subgroups classified according to the stage at which BCPs are stalled in differentiation and cytogenetic/molecular markers.1 Importantly, different biological subtypes of B-ALL have distinct causal mechanisms and show different clinical outcomes.1-3
High hyperdiploid (HyperD) B-ALL (HyperD-ALL) is the most common subtype of childhood B-ALL and is characterized by the presence of 51 to 67 chromosomes in leukemic cells.4-7 HyperD-ALL comprises ∼30% of pediatric B-ALL and usually has a favorable clinical outcome.5,8,9 Of note, the distribution of chromosome gains is not random and preferentially shows gains of chromosomes X, 4, 6, 10, 14, 17, 18, and 21.5,10 Hyperdiploidy is an initiating oncogenic event in B-ALL and secondary alterations necessary for clinical B-ALL accumulate subclonally and postnatally.5,7,11,12 Despite hyperdiploidy being the most common B-ALL in children, very little is known about its etiology and pathogenesis, and many questions about the biology of HyperD-ALL remain unanswered. Despite its favorable clinical outcome, precise knowledge of the physiopathogenic mechanisms underlying HyperD-ALL is necessary because, in absolute numbers, the morbidity/mortality associated with HyperD-ALL still represent a clinical challenge.
HyperD-ALL is proposed to arise in a BCP in utero.7 However, the causal molecular mechanisms of hyperdiploidy in BCPs remain elusive. In this sense, faithful chromosome segregation is essential for maintaining the genomic integrity of eukaryotic cells, and deficient chromosome segregation leads to aneuploidy and cancer.13-15 Three main and nonmutually exclusive mechanisms, interconnected with mitosis,16 underlie chromosome missegregation: (i) defects in bipolar spindle formation, (ii) defects in chromosome structure and function, and (iii) defects in the spindle assembly checkpoint (SAC), which controls proper mitosis until chromosomes are properly attached to the spindle.14-16 Indeed, SAC defects have been proposed to be an underlying pathogenic mechanism in rare cases of ETV6/RUNX1+ B-ALL with near-tetraploid karyotypes.17 Therefore, abnormal mitotic control in BCP could be at the origin of hyperdiploidy in B-ALL.
Here, we used a large cohort of primary pediatric B-ALL samples (n = 54) to gain insights into the cellular and molecular mechanisms underlying mitotic/chromosome defects predicated to be at the origin of pediatric HyperD-ALL. Our data reveal that HyperD-ALL blasts show robust condensin-complex defects and defective Aurora B kinase (AURKB) activity, leading to abnormal mitotic progression and chromosome missegregation. Functional inhibition of AURKB and the SAC in normal hematopoietic stem/progenitor cells (HSPCs) reproduced hyperdiploid karyotypes with abnormal chromosome structure. We conclude that defects in the condensin complex, AURKB, and SAC are associated with HyperD-ALL, likely representing a pathogenic mechanism.
Methods
Pediatric B-ALL leukemic samples and cell lines
Diagnostic BM samples from B-ALL pediatric patients were obtained from collaborating hospitals. B-ALL diagnosis was based on French-American-British (FAB) and World Health Organization (WHO) classifications.1 Supplemental Table 1 (available on the Blood Web site) summarizes main clinico-biological data of the patients. Fetal tissue was collected from developing embryos aborted at 18 to 22 weeks of pregnancy, obtained from the Medical Research Council (MRC)/Wellcome Trust Human Developmental Biology Resource upon informed consent and approval by our local ethics committee. The B-ALL cell lines SEM, REH, and MHH-CALL-2 (German Collection of Microorganisms and Cell Cultures [DMSZ], Braunschweig, Germany) were used for confirmatory studies. This study was approved by the Barcelona Clínic-Hospital Institutional Review Ethics Board (HCB/2014/0687), and patient samples were accessed upon informed consent.
In vivo expansion of B-ALL blasts
All experimental procedures were approved by the Animal Care Committee of the Barcelona Biomedical Research Park (DAAM7393). Primary blasts (5 × 105) were expanded in vivo in sublethally irradiated 8- to 14-week-old nonobese diabetic/LtSz-scid IL-2Ry−/− mice (NSG) upon intra-BM transplantation.18 Peripheral blood (PB) was monitored by fluorescence-activated cell sorting (FACS) for leukemia engraftment. Primografts were sacrificed when engraftment reached 10% to 15% in PB, typically representing >80% engraftment in BM. Blasts were isolated from BM and spleen by density-gradient centrifugation for downstream analysis. For FACS analysis of leukemic engraftment, PB mononuclear cells were stained with anti-human HLA-ABC fluorescein isothiocyanate (FITC), CD19-phycoerythrin, and CD45-allophycocyanin antibodies (BD Biosciences), and analyzed using a FACSCantoII cytometer.
Indirect immunofluorescence
B-ALL cells were spun on poly-l-lysine-coated coverslips (500g, 3 minutes) before fixation. Cells were fixed and permeabilized with Triton X-100–containing buffer (5 minutes). Cells were blocked with permeabilization buffer containing 1% to 3% bovine serum albumin (1 hour, 37°C), and incubated overnight at 4°C with primary antibodies (supplemental Table 2). Cells were washed with permeabilization buffer, and incubated (45 minutes) with fluorophore-conjugated secondary antibodies (The Jackson Laboratory). All antibodies were diluted in blocking buffer. Slides were mounted with Vectashield 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Details of chromosome spreading for immunofluorescence are provided in supplemental Methods.
Confocal microscopy and image acquisition
Microscope images were captured using a fully equipped Zeiss LSM880 laser-scanning spectral confocal microscope equipped with an AxioObserver Z1 inverted microscope. DAPI, Alexa 488, Alexa Fluor 555, and Alexa Fluor 647 images were acquired sequentially using 405, 488, 561, and 633 lasers, dichroic beam splitters, emission detection ranges of 415 to 480 nm, 500 to 550 nm, 571 to 625 nm, and 643 to 680 nm, respectively, and the confocal pinhole was set at 1 Airy unit (AU). An acoustic optical-beam splitter was used at the same emission detection ranges. Spectral detection was performed using 2 photomultipliers and 1 central GaAsP detector used for the acquisition of Alexa 647. Images were acquired in a 1024- × 300-pixel format, zoom was set at 2, pixel size at 53 × 53 nm, and dwell time at 0.51 microseconds. Z-stacks were acquired at a 300-nm step size to reconstruct the entire nuclei volume. Immunofluorescence signal quantification was performed using FIJI-ImageJ (NIH). Details of image quantifications are provided in supplemental Methods.
Details for in vitro culture/expansion of B-ALL cells and HSPCs, chromosome function and cytogenetic analysis, fluorescence quantification, western blot (WB) and protein analysis, mass-spectometry (MS) assays, cell cycle and phosphor-H3S10 quantification, reverse transcription–polymerase chain reaction (PCR), chromatin immunoprecipitation, and RNA sequencing (RNA-seq) are provided in supplemental Methods and supplemental Tables 3 and 4.
Statistical analysis
Statistical comparisons were performed using GraphPad Prism. Mean values and their standard error of the mean were calculated for each variable. All data were analyzed according to the test indicated in the appropriate figure legends on the indicated number of experiments. Non-HyperD were compared with HyperD B-ALLs. A value of P < .05 was considered statistically significant.
Results
HyperD-ALL cells are low proliferative and show a delay in early mitosis
Aneuploid cells typically display a gene signature characterized by an upregulation of genes involved in oxidative stress response, membrane functions, and immune response regulation coupled with a downregulation of genes involved in cell proliferation and nucleic acid metabolism.19-21 We first analyzed the transcriptomic signature of HyperD-ALL blasts using an RNA-seq data set from HyperD-ALL patients (n = 58) and non-HyperD B-ALL patients (n = 30, nonaneuploid; supplemental Methods).22 A gene ontology analysis of the 26 239 genes differentially expressed between primary HyperD-ALL and non-HyperD B-ALLs confirmed an aneuploidy-like gene expression signature characterized by the upregulation of pathways associated with oxidative stress, protein turnover, cell death, immune system activation, and membrane functions (Figure 1A-B), and downregulation of pathways associated with nucleic acid metabolism and transfer RNA biology (Figure 1A,C). Consistent with this, we found that the aneuploid pediatric B-ALL cell line CALL-2 (doubled-up hypodiploid karyotype: 51XX,+X,+18,+der(18)t(15;18),+21,+21)23 exhibits a significantly lower proliferative rate than the non-HyperD-ALL cell lines SEM and REH (Figure 1D). Mechanistically, CALL-2 cells revealed a fivefold increase in apoptosis coupled to an accumulation in G2/M (Figure 1E-F), indicating cell division/mitotic defects in aneuploid B-ALL cells.
![HyperD-ALL cells are low proliferative and show a delay in early mitosis. (A) Heat map of the top 50 genes more differentially expressed between HyperD (n = 58) and non-HyperD (n = 30) B-ALL samples. (B-C) Top 20 statistically significant upregulated (B) or downregulated (C) biological pathways identified using GSEA for the genes differentially expressed in HyperD vs nonHyperD-ALL patients. Colored bars represent normalized enrichment scores (NES). P values are shown. (D) Sixteen-day proliferation curves for the indicated cell lines; n = 3 independent experiments. (E) SubG0/SubG1 apoptotic levels identified by FACS for the indicated cell lines; n = 3 independent experiments. (F) Cell cycle analysis for the indicated cell lines. Left, representative cell cycle FACS analysis. Right, frequency of cells in G2/M analyzed; n = 3 independent experiments. (G) Representative DNA-Kinetochore-spindle IF staining (DNA, ACA, tubulin, and pericentrin) identifying the different mitotic phases in B-ALL cell lines. The SAC identifies the transition from early to late mitosis. Scale bar, 10 µm. (H-I) Mitosis progression in B-ALL cell lines. Progression from early to late mitosis (H), and frequency of cells at the indicated mitotic phases (I); n = 4 independent experiments. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01 (2-way analysis of variance [ANOVA]). A, anaphase; CK, cytokinesis; M, metaphase; P, prophase; PM, prometaphase; T, telophase.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/3/10.1182_blood.2019002538/1/m_bloodbld2019002538f1.png?Expires=1770500385&Signature=VQPxfGa6rce7uUMLTOHc8-GbH2rhiqNPhUEfCehKccwTxU-DrDh0oZd1McGMPluDrnB1kWhUHuBRwZuq6t9F0KXNZ1pjpp7N8mg91TmPKBOxkujztuOCF50GxekPYpxtCSYlJfEmU6zdXBVbweGRy5jP80MROO9aOqa~i~DLCGXbWyeK0yBAyHDnOYrfqXFUo8urmZck5Tq2lO3Bb~-hMM~0aPvsV1g-V8t8W2EccikmYJCEkrfd9frA4XuACz8TJnLLHjgVZ72mivYgM74rlbsVwGL7kNJhpiLlaBYZCj13qol8LeO8Ppk8sI5ZhAktB8K2cUtT4BNrEmrWSEVP8Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HyperD-ALL cells are low proliferative and show a delay in early mitosis. (A) Heat map of the top 50 genes more differentially expressed between HyperD (n = 58) and non-HyperD (n = 30) B-ALL samples. (B-C) Top 20 statistically significant upregulated (B) or downregulated (C) biological pathways identified using GSEA for the genes differentially expressed in HyperD vs nonHyperD-ALL patients. Colored bars represent normalized enrichment scores (NES). P values are shown. (D) Sixteen-day proliferation curves for the indicated cell lines; n = 3 independent experiments. (E) SubG0/SubG1 apoptotic levels identified by FACS for the indicated cell lines; n = 3 independent experiments. (F) Cell cycle analysis for the indicated cell lines. Left, representative cell cycle FACS analysis. Right, frequency of cells in G2/M analyzed; n = 3 independent experiments. (G) Representative DNA-Kinetochore-spindle IF staining (DNA, ACA, tubulin, and pericentrin) identifying the different mitotic phases in B-ALL cell lines. The SAC identifies the transition from early to late mitosis. Scale bar, 10 µm. (H-I) Mitosis progression in B-ALL cell lines. Progression from early to late mitosis (H), and frequency of cells at the indicated mitotic phases (I); n = 4 independent experiments. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01 (2-way analysis of variance [ANOVA]). A, anaphase; CK, cytokinesis; M, metaphase; P, prophase; PM, prometaphase; T, telophase.
HyperD-ALL cells are low proliferative and show a delay in early mitosis. (A) Heat map of the top 50 genes more differentially expressed between HyperD (n = 58) and non-HyperD (n = 30) B-ALL samples. (B-C) Top 20 statistically significant upregulated (B) or downregulated (C) biological pathways identified using GSEA for the genes differentially expressed in HyperD vs nonHyperD-ALL patients. Colored bars represent normalized enrichment scores (NES). P values are shown. (D) Sixteen-day proliferation curves for the indicated cell lines; n = 3 independent experiments. (E) SubG0/SubG1 apoptotic levels identified by FACS for the indicated cell lines; n = 3 independent experiments. (F) Cell cycle analysis for the indicated cell lines. Left, representative cell cycle FACS analysis. Right, frequency of cells in G2/M analyzed; n = 3 independent experiments. (G) Representative DNA-Kinetochore-spindle IF staining (DNA, ACA, tubulin, and pericentrin) identifying the different mitotic phases in B-ALL cell lines. The SAC identifies the transition from early to late mitosis. Scale bar, 10 µm. (H-I) Mitosis progression in B-ALL cell lines. Progression from early to late mitosis (H), and frequency of cells at the indicated mitotic phases (I); n = 4 independent experiments. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01 (2-way analysis of variance [ANOVA]). A, anaphase; CK, cytokinesis; M, metaphase; P, prophase; PM, prometaphase; T, telophase.
To further characterize the mitotic progression of HyperD-ALL cells, we stained CALL-2, SEM, and REH for DAPI, tubulin, pericentrin, and the anti-centromere antibody (ACA) to unequivocally identify the different mitotic phases24 (Figure 1G). Consistent with the FACS data, immunofluorescence (IF) analysis revealed that CALL-2 cells accumulated in early mitosis, specifically at prometaphase/metaphase, with a concomitant delay in late mitosis (Figure 1H-I).
Chromosome-alignment defects in prometaphase underlie the mitotic delay of HyperD-ALL primary blasts
Cancer cell lines do not faithfully phenocopy the molecular complexity of the disease. Furthermore, despite CALL-2 being the only childhood HyperD-ALL cell line available, it actually represents a doubled-up hypodiploid B-ALL cell line.23 We thus aimed to analyze the proliferation/mitotic defects in childhood HyperD-ALL primary blasts. Because leukemic primary cells fail to expand ex vivo,25-27 we cocultured B-ALL blasts with Nestin-positive fetal BM (FBM)-derived mesenchymal stem cells (MSCs), which support short-term proliferation of primary B-ALL blasts without compromising clonal composition28 (Figure 2A; supplemental Figure 1a-b). Similar to cell lines, non-HyperD-ALL primary cells grown on Nestin-positive FBM-MSCs expanded fivefold over a 6-day period whereas HyperD-ALL primary blasts failed to expand at all ex vivo (Figure 2B). FACS analysis using BM-diagnostic samples also showed an increase in both apoptosis and frequency of G2/M cells in primary HyperD-ALL cells (Figure 2C-D). To further link proliferative impairment with mitotic defects, we analyzed the mitotic progression in dividing B-ALL primary blasts expanded in vivo in NSG mice (Figure 2E; supplemental Figure 2a). Xenografted blasts were processed and IF-stained for the DNA-Kinetochore-Spindle staining to unambiguously identify each mitotic phase (Figure 2F). Consistent with cell lines, HyperD-ALL primary blasts accumulated in early mitosis, at prometaphase/metaphase, with a concomitant delay in late mitosis (telophase and cytokinesis; Figure 2G; supplemental Figure 2b).

HyperD-ALL primary blasts show a delay in early mitosis associated with chromosome-alignment defects in prometaphase. (A) Schematic depicting the experimental design for ex vivo growth of primary B-ALL blasts onto Nestin-positive hBM-MSCs. (B) Left, representative images of primary non-HyperD and HyperD-B-ALL ex vivo cultures on Nestin-positive hBM-MSCs at the indicated time points. Right, absolute counts of B-ALL primary blasts at the indicated time points; n = 2. (C-D) Frequency of apoptotic (SubG0/SubG1) (C) and G2/M (D) non-HyperD and HyperD-ALL primary cells from BM samples; n = 3 patients of each. (E) Schematic depicting the PDX model used to expand primary B-ALL blasts in vivo. (F) Representative DNA (blue)-Kinetochore (purple)-spindle (red-green) IF staining identifying the different mitotic phases in PDX-expanded B-ALLs. The SAC identifies the transition from early to late mitosis. (G) Mitosis progression of PDX-expanded B-ALL primary cells. Left, progression from early to late mitosis. Right, frequency of cells at the indicated mitotic phases; n = 3 non-HyperD and n = 5 HyperD PDX-expanded B-ALLs. (H) Left, representative images of mitotic cells with nonaligned and aligned chromosomes at the metaphase plate. Right, frequency of PDX-expanded B-ALL primary blasts showing chromosome alignment at prometaphase/metaphase; n = 4 non-HyperD and n = 4 HyperD PDX-expanded B-ALLs. (I) Schematic depicting the chromosome-biorientation assay. (J) Representative images of the DNA-Kinetochore-spindle staining in monastrol/MG132-treated cells with 0 (left), 1 (middle), and >2 (right) misaligned chromosomes. (K) Quantification of metaphase cells showing misaligned chromosomes; n = 3 non-HyperD and n = 3 HyperD PDX-expanded B-ALLs. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .0001 (2-way ANOVA). Scale bars, 10 µm.
HyperD-ALL primary blasts show a delay in early mitosis associated with chromosome-alignment defects in prometaphase. (A) Schematic depicting the experimental design for ex vivo growth of primary B-ALL blasts onto Nestin-positive hBM-MSCs. (B) Left, representative images of primary non-HyperD and HyperD-B-ALL ex vivo cultures on Nestin-positive hBM-MSCs at the indicated time points. Right, absolute counts of B-ALL primary blasts at the indicated time points; n = 2. (C-D) Frequency of apoptotic (SubG0/SubG1) (C) and G2/M (D) non-HyperD and HyperD-ALL primary cells from BM samples; n = 3 patients of each. (E) Schematic depicting the PDX model used to expand primary B-ALL blasts in vivo. (F) Representative DNA (blue)-Kinetochore (purple)-spindle (red-green) IF staining identifying the different mitotic phases in PDX-expanded B-ALLs. The SAC identifies the transition from early to late mitosis. (G) Mitosis progression of PDX-expanded B-ALL primary cells. Left, progression from early to late mitosis. Right, frequency of cells at the indicated mitotic phases; n = 3 non-HyperD and n = 5 HyperD PDX-expanded B-ALLs. (H) Left, representative images of mitotic cells with nonaligned and aligned chromosomes at the metaphase plate. Right, frequency of PDX-expanded B-ALL primary blasts showing chromosome alignment at prometaphase/metaphase; n = 4 non-HyperD and n = 4 HyperD PDX-expanded B-ALLs. (I) Schematic depicting the chromosome-biorientation assay. (J) Representative images of the DNA-Kinetochore-spindle staining in monastrol/MG132-treated cells with 0 (left), 1 (middle), and >2 (right) misaligned chromosomes. (K) Quantification of metaphase cells showing misaligned chromosomes; n = 3 non-HyperD and n = 3 HyperD PDX-expanded B-ALLs. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .0001 (2-way ANOVA). Scale bars, 10 µm.
Because chromosomes align at the metaphase plate in the prometaphase-to-metaphase transition, we then inspected dividing cells in early mitosis to distinguish between prometaphase (nonaligned chromosomes) and metaphase (aligned chromosomes) cells. We observed a robust decrease of HyperD-ALL blasts with aligned metaphase plates (Figure 2H), confirming chromosome-alignment defects in early mitosis in HyperD-ALL cells. Chromosome alignment relies on the dynamics of chromosome biorientation to the spindle poles.29 We thus analyzed the dynamics of chromosome biorientation in primary B-ALL blasts by generating monopolar spindles and prometaphase arrest with monastrol (a spindle bipolarity inhibitor), followed by treatment with the proteasome inhibitor MG132, which further arrests dividing cells in metaphase30 (Figure 2I). Strikingly, non-HyperD-ALL primary blasts properly aligned chromosomes at the metaphase plate in ∼90% of the metaphases (Figure 2J-K). However, HyperD-ALL samples showed a massive decrease in the number of cells with aligned chromosomes at the metaphase plate (∼45%) (Figure 2J-K). It is noteworthy that the defective chromosome biorientation in HyperD-ALL cells was confirmed in B-ALL cell lines (supplemental Figure 3a). Thus, the impaired proliferation of HyperD-ALL blasts may result from mitotic defects in prometaphase-metaphase due to aberrant chromosome alignment at the metaphase plate.
Chromosome-misalignment defects result in chromosome missegregation and nonmodal karyotypes in HyperD-ALL blasts
Because defects in chromosome alignment often result in chromosome missegregation,24 we next analyzed the rates of chromosome-segregation defects, mainly lagging chromosomes and anaphase bridges, in B-ALL blasts (Figure 3A). To overcome the accumulation of HyperD-ALL cells in early mitosis, B-ALL primary blasts were treated with the cytokinesis inhibitor Blebbistatin.31 We found that HyperD-ALL primary blasts displayed a significantly, approximately fourfold, higher number of late mitosis with chromosome-segregation defects (Figure 3B).
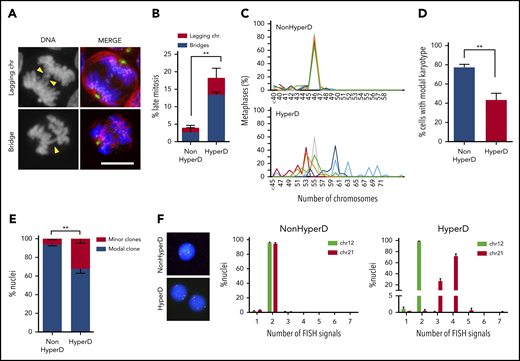
Chromosome-segregation defects and nonmodal karyotypes in HyperD-ALL blasts. (A) Representative DNA (blue)-Kinetochore (purple)-spindle (green and red) IF staining identifying lagging and bridge chromosomes. Yellow arrowheads depict the indicated chromosome-segregation defect. (B) Frequency of blebbistatin-treated mitotic PDX-expanded primary blasts with lagging and bridge chromosomes; n = 151 mitosis from 3 non-HyperD and 96 mitosis from 3 HyperD-ALLs. (C) Comparison of modal karyotypes from 50 metaphases from primary HyperD (n = 6) and non-HyperD (n = 6) B-ALL samples. (D) Frequency of cells showing modal karyotype. (E-F) FISH analysis using DNA probes for chromosomes 12 (green) and 21 (red) of 200 interphase nuclei from n = 3 non-HyperD and 4 HyperD-ALL primary samples. (E) Frequency of cells representing the modal clone vs minor clones. (F) Representative FISH analysis for a primary non-HyperD and a HyperD-ALL. Graphs represent the mean and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .0001 (2-way ANOVA). Scale bars, 10 µm.
Chromosome-segregation defects and nonmodal karyotypes in HyperD-ALL blasts. (A) Representative DNA (blue)-Kinetochore (purple)-spindle (green and red) IF staining identifying lagging and bridge chromosomes. Yellow arrowheads depict the indicated chromosome-segregation defect. (B) Frequency of blebbistatin-treated mitotic PDX-expanded primary blasts with lagging and bridge chromosomes; n = 151 mitosis from 3 non-HyperD and 96 mitosis from 3 HyperD-ALLs. (C) Comparison of modal karyotypes from 50 metaphases from primary HyperD (n = 6) and non-HyperD (n = 6) B-ALL samples. (D) Frequency of cells showing modal karyotype. (E-F) FISH analysis using DNA probes for chromosomes 12 (green) and 21 (red) of 200 interphase nuclei from n = 3 non-HyperD and 4 HyperD-ALL primary samples. (E) Frequency of cells representing the modal clone vs minor clones. (F) Representative FISH analysis for a primary non-HyperD and a HyperD-ALL. Graphs represent the mean and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .0001 (2-way ANOVA). Scale bars, 10 µm.
Because chromosome missegregation leads to aneuploidy, we analyzed the modal karyotype distribution in 12 diagnostic B-ALL samples. We found high chromosome stability in non-HyperD B-ALL samples, with a modal chromosome number of 46 in >80% of the metaphases analyzed (Figure 3C-D; supplemental Figure 4). However, HyperD-ALL blasts showed an increased karyotype instability defined by the presence of a major clone (40% of the cells) and minor clones with nonmodal chromosome distributions (Figure 3C-D; supplemental Figure 4). These results were confirmed by fluorescence in situ hybridization (FISH) analysis for chromosomes 12 and 21 (Figure 3E-F). Chromosome instability in HyperD-ALL cells was further confirmed in B-ALL cell lines (supplemental Figure 3b-c). Collectively, chromosome-alignment defects result in chromosome-segregation defects and subsequent nonmodal karyotypes in HyperD-ALL blasts.
HyperD-ALL blasts show chromosome hypocondensation and loss of centromere stiffness due to condensin-complex defects
We next investigated the mechanisms leading to these mitotic/chromosome defects. IF analysis of the spindle using the DNA-Kinetochore-Spindle staining showed similarly low frequency of mitotic blasts with spindle abnormalities such as multipolar or disorganized spindles in non-HyperD and HyperD-ALL (Figure 4A). Moreover, defects in bipolar spindle formation frequently lead to cytokinesis defects and tetraploidization.32 Because no (near)-tetraploid cells were observed in any HyperD-ALL patient (Figure 3C-D; supplemental Figure 4), we ruled out defects in spindle formation in HyperD-ALL blasts.
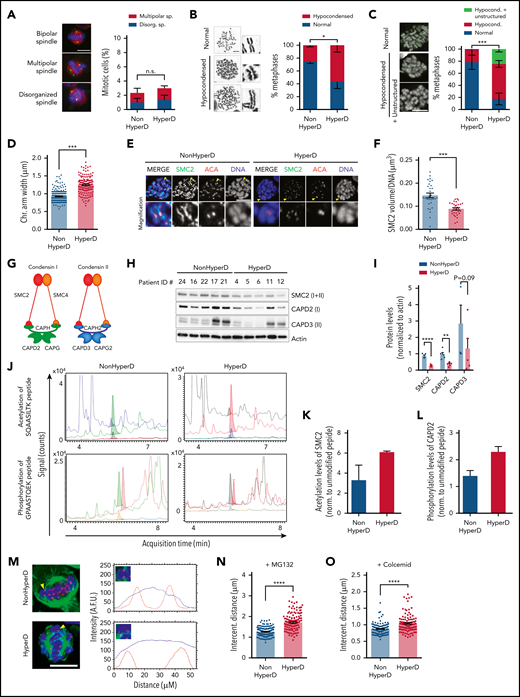
HyperD-ALL blasts show chromosome hypocondensation, loss of centromere stiffness, and defects in the condensin complex. (A) Analysis of spindle abnormalities in B-ALL primary blasts. Left, representative DNA-Kinetochore-spindle IF staining of mitotic cells with bipolar, multipolar, and disorganized spindles. Right, frequency of mitotic cells displaying spindle defects; n = 3 non-HyperD-ALLs (n = 251 mitosis) and n = 3 HyperD-ALLs (n = 251 mitosis) PDX-expanded samples. (B) Frequency of metaphases with hypocondensed chromosomes in primary B-ALL blasts; n = 200 metaphases from 4 non-HyperD and n = 250 metaphases from 5 HyperD-ALLs primary samples. Left, representative images of normal and hypocondensed metaphase chromosomes. Insets represent ×3 magnifications. (C) Chromosome structure of formaldehyde-crosslinked PDX-expanded B-ALL samples. Left, Representative images of metaphase cells with hypocondensed chromosomes. Anti-ACA staining is shown in green. Right, frequency of formaldehyde-crosslinked metaphases showing hypocondensed or hypocondensed with unstructured chromosomes in B-ALL primary samples; n = 60 metaphases from 3 non-HyperD and n = 57 metaphases from 3 HyperD-ALLs. (D) Chromosome arm width using PDX-expanded B-ALL samples from panel C; n = 191 chromosomes from 3 non-HyperD and n = 143 chromosomes from 3 HyperD-ALLs. (E) Representative IF images of metaphase PDX-expanded B-ALL blasts stained with DAPI, anti-SMC2, and anti-ACA. (F) Quantification of the SMC2 total volume in metaphase chromosomes from panel E; n = 30 metaphases from 3 non-HyperD and 3 HyperD-ALLs. (G) Schematic cartoon of the 2 human condensin complexes. (H) WB analysis of the indicated condensin members in whole-cell lysates from PDX-expanded B-ALL samples. (I) Quantification of WB bands from panel H normalized to actin. (J) Representative HPLC-ESI-MS chromatograms of the indicated peptides for HyperD and non-HyperD PDX-expanded B-ALLs. (K) Acetylation levels of SMC2 peptide SQAASILTK (m/z = 480.8). (L) Phosphorylation levels of CAPD2 peptide GPAASTQEK (m/z = 524.7). Results depict the average of the peak areas from independent MS experiments from 2 non-HyperD and 2 HyperD-ALL PDX-expanded blasts. (M) Representative line-scan measurements of individual centromeres in the indicated B-ALL primary samples. DAPI and ACA are depicted as a blue and red lines, respectively. Yellow arrowheads point to the analyzed chromosome. (N) Intercentromeric distance from MG132-treated PDX-expanded B-ALL blasts; n = 155 centromeres from 3 non-HyperD and n = 119 centromeres from 3 HyperD-ALLs. (O) Intercentromeric distance from colcemid-treated PDX-expanded B-ALL blasts; n = 130 centromeres from 3 non-HyperD and n = 111 centromeres from 3 HyperD-ALLs. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .001; ****P < .0001. Two-way ANOVA (A-C) or Student t test (D, F, I, N, O). Scale bars, 10 µm.
HyperD-ALL blasts show chromosome hypocondensation, loss of centromere stiffness, and defects in the condensin complex. (A) Analysis of spindle abnormalities in B-ALL primary blasts. Left, representative DNA-Kinetochore-spindle IF staining of mitotic cells with bipolar, multipolar, and disorganized spindles. Right, frequency of mitotic cells displaying spindle defects; n = 3 non-HyperD-ALLs (n = 251 mitosis) and n = 3 HyperD-ALLs (n = 251 mitosis) PDX-expanded samples. (B) Frequency of metaphases with hypocondensed chromosomes in primary B-ALL blasts; n = 200 metaphases from 4 non-HyperD and n = 250 metaphases from 5 HyperD-ALLs primary samples. Left, representative images of normal and hypocondensed metaphase chromosomes. Insets represent ×3 magnifications. (C) Chromosome structure of formaldehyde-crosslinked PDX-expanded B-ALL samples. Left, Representative images of metaphase cells with hypocondensed chromosomes. Anti-ACA staining is shown in green. Right, frequency of formaldehyde-crosslinked metaphases showing hypocondensed or hypocondensed with unstructured chromosomes in B-ALL primary samples; n = 60 metaphases from 3 non-HyperD and n = 57 metaphases from 3 HyperD-ALLs. (D) Chromosome arm width using PDX-expanded B-ALL samples from panel C; n = 191 chromosomes from 3 non-HyperD and n = 143 chromosomes from 3 HyperD-ALLs. (E) Representative IF images of metaphase PDX-expanded B-ALL blasts stained with DAPI, anti-SMC2, and anti-ACA. (F) Quantification of the SMC2 total volume in metaphase chromosomes from panel E; n = 30 metaphases from 3 non-HyperD and 3 HyperD-ALLs. (G) Schematic cartoon of the 2 human condensin complexes. (H) WB analysis of the indicated condensin members in whole-cell lysates from PDX-expanded B-ALL samples. (I) Quantification of WB bands from panel H normalized to actin. (J) Representative HPLC-ESI-MS chromatograms of the indicated peptides for HyperD and non-HyperD PDX-expanded B-ALLs. (K) Acetylation levels of SMC2 peptide SQAASILTK (m/z = 480.8). (L) Phosphorylation levels of CAPD2 peptide GPAASTQEK (m/z = 524.7). Results depict the average of the peak areas from independent MS experiments from 2 non-HyperD and 2 HyperD-ALL PDX-expanded blasts. (M) Representative line-scan measurements of individual centromeres in the indicated B-ALL primary samples. DAPI and ACA are depicted as a blue and red lines, respectively. Yellow arrowheads point to the analyzed chromosome. (N) Intercentromeric distance from MG132-treated PDX-expanded B-ALL blasts; n = 155 centromeres from 3 non-HyperD and n = 119 centromeres from 3 HyperD-ALLs. (O) Intercentromeric distance from colcemid-treated PDX-expanded B-ALL blasts; n = 130 centromeres from 3 non-HyperD and n = 111 centromeres from 3 HyperD-ALLs. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .001; ****P < .0001. Two-way ANOVA (A-C) or Student t test (D, F, I, N, O). Scale bars, 10 µm.
We next assessed whether chromosome structure and function underlie the mitotic defects observed in HyperD-ALL. We first examined the chromosome morphology of Carnoy-fixed pediatric B-ALL blasts (supplemental Table 1). Although non-HyperD-ALL metaphases mostly showed normal rod-shaped chromosomes, ∼60% of HyperD-ALL metaphases displayed curly shaped/hypocondensed chromosomes with irregular borders (Figure 4B; supplemental Figure 5a). Importantly, intrapatient comparison of diploid normal hematopoietic cells vs HyperD blasts confirmed that chromosome hypocondensation specifically occurs in HyperD-ALL cells (10% vs 70%; supplemental Figure 5b). Chromosome hypocondensation defects were further confirmed in B-ALL cell lines (supplemental Figure 5c). We next scored formaldehyde-fixed B-ALL samples for chromosome structure/condensation, and found significantly more metaphases containing hypocondensed and unstructured fuzzier and wider chromosomes in HyperD-ALL than in non-HyperD-ALL blasts (80% vs 20%; P < .001; Figure 4C-D; supplemental Figure 5d), excluding an impact of the fixative on chromosome-structure/condensation defects.
Condensin complexes are major components of the chromosome scaffold that regulate chromosome compaction and higher-order chromatin organization during mitosis.33-36 The chromosome-structure/condensation defects observed in HyperD-ALLs prompted us to analyze in chromosome spreads from primary B-ALL samples the binding pattern of SMC2, a major component of condensin complexes. Non-HyperD-ALL blasts showed a normal beaded pattern for SMC2, spreading along the chromatids with a centromere enrichment (Figure 4E). In contrast, SMC2 was hardly detectable in neither chromatids nor centromeres, and showed an abnormal staining pattern in HyperD-ALL blasts (Figure 4E). Indeed, 3-dimensional image quantification of SMC2 staining revealed a significantly lower volume of SMC2 in chromosomes from HyperD-ALL blasts (Figure 4F; supplemental Figure 5e).
To further characterize the defects in condensin complexes in HyperD-ALL samples, we analyzed by WB in primary-derived xenograft (PDX)–expanded B-ALL cells the distinct protein members specific for each of the 2 human condensin complexes (complex I and II), which play a differential contribution to mitotic chromosome organization/segregation (Figure 4G). Protein analysis confirmed the lower levels of SMC2 in HyperD-ALL blasts, and revealed that both condensin complexes (CAPD2 and CAPD3) were similarly affected (Figure 4H-I). Strikingly, however, no differences were observed at the RNA level for any of the condensin-complex members between HyperD-ALL and non-HyperD-ALL samples (supplemental Figure 5f), suggesting that posttranslational modifications (PTMs) may underlie condensin-complex defects in HyperD-ALL blasts.
Compelling data strongly suggest that PTMs are essential for regulating condensin loading to chromosomes.37,38 We thus performed MS analyses for both SMC2 acetylation and CAPD2 phosphorylation levels after immunoprecipitation of the condensin complexes with anti-SMC2 (supplemental Figure 5g), and found increased levels of both SMC2 acetylation and CAPD2 phosphorylation in HyperD-ALL samples (Figure 4J-L), suggesting that PTMs regulating condensin activation may represent a mechanism underlying condensin defects in HyperD-ALL blasts.
Condensin complexes set the stiffness of the centromeric chromatin required to withstand the spindle-pulling forces during metaphase,29 and SMC2 depletion results in increased intercentromeric distances in metaphase chromosomes.39,40 To analyze centromere stiffness in HyperD-ALL blasts, we measured the intercentromeric distances between sister-kinetochore pairs in metaphase-arrested blasts (Figure 4M). For this, B-ALL blasts were metaphase-arrested with chromosomes under tension (nonrelaxed length) or without tension (rest length) from the spindle, by using either the proteasome inhibitor MG132 or the microtubule depolymerizer colcemid, respectively. HyperD-ALL blasts consistently displayed a significantly longer intercentromeric distance than non-HyperD-ALL blasts (Figure 4N-O). Collectively, PTMs of condensin members may induce a defective condensin complex that leads to high-order chromosome-organization defects and impaired centromere stiffness/stretching at metaphase in HyperD-ALL blasts.
Kinetochores are normal in HyperD-ALL blasts
The kinetochore binds microtubules at centromeres and regulates chromosome segregation.41 CENP-A (centromere-specific histone H3) and NDC80/NUF-2 are key centromere chromatin markers of the inner and outer kinetochore plate, respectively, which control kinetochore’s assembly. CENP-A overlaps with the condensin complex and it is flanked by the heterochromatin histone marks H3K9me3 and H3K27me3.42-47 We thus were prompted to study whether the condensin defects observed in HyperD-ALL blasts are associated with centromeric chromatin defects and destabilization of the kinetochore. Chromatin immunoprecipitation–quantitative PCR assays showed no differences of CENP-A, H3K9me3, or H3K27me3 levels at centromeres of HyperD-ALL blasts as compared with non-HyperD-ALL blasts (supplemental Figure 6a). Moreover, quantitative confocal microscopy analysis revealed very similar levels of NUF-2 between HyperD-ALL and non-HyperD-ALL blasts (supplemental Figure 6b-c), indicating that despite impaired centromere stiffness, the centrochromatin is not epigenetically impaired and the kinetochore forms normally in HyperD-ALL blasts.
The CPC proteins AURKB and Survivin are mislocalized from the inner centromere in early mitotic HyperD-ALL blasts
We next aimed to analyze the cellular mechanisms by which the impaired condensin complex leads to mitotic/chromosome defects in HyperD-ALL blasts. We first analyzed the chromosome passenger complex (CPC), a protein complex composed of AURKB and the accessory subunits Survivin, Borealin, and INCENP. The CPC regulates the SAC to ensure proper kinetochore-microtubule attachment, and was shown mislocalized from the inner centromere in cells with a defective condensin complex.34,48,49 Prometaphase-arrested non-HyperD-ALL primary blasts revealed a normal chromosomal distribution of both AURKB and Survivin mainly concentrated in the inner centromere (Figure 5A-B). However, HyperD-ALL blasts showed an aberrant chromosomal distribution of both AURKB and Survivin, diffusely distributed throughout the chromosome arms rather than concentrated in the inner centromere (Figure 5A-B). Quantification of both AURKB and Survivin at the inner centromeres confirmed a significant decrease of both CPC proteins in centromeres of HyperD-ALL blasts (Figure 5C-D; supplemental Figure 7a-b). To further characterize the localization of AURKB and Survivin, we analyzed the frequency of chromosomes showing either centromeric or scattered localization (Figure 5E), and confirmed that HyperD-ALL blasts preferentially showed scattered localization throughout the chromosome arms (Figure 5F-G). These results were reproduced using B-ALL cell lines (supplemental Figure 7c-d). Of note, the overall chromosome-wide expression levels of both AURKB and phosphohistone H3 at serine 10 (H3S10p), the major readout of AURKB activity, were significantly reduced in HyperD-ALL blasts (Figure 5H-I). Finally, we generated condensin complex I–defective non-HyperD-ALL cell lines by knocking down CAPD2, and confirmed the mislocalization of AURKB from the inner centromere (supplemental Figure 7e-f). Taken together, mislocalization of the CPC proteins AURKB and Survivin in early mitosis represents a major mechanism linking defective condensin complex and chromosome-alignment/segregation defects in HyperD-ALL blasts.
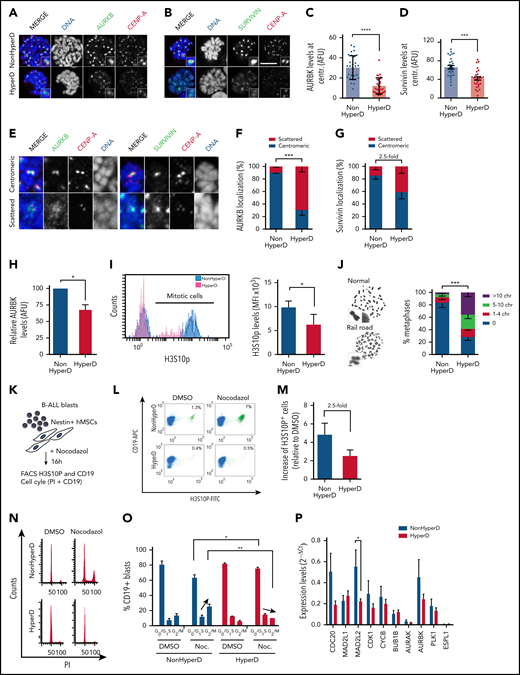
Mislocalized AURKB and Survivin from the inner centromere and loss of chromatid cohesion and SAC impairment in HyperD-ALL blasts. (A-B) Representative IF staining for CENP-A and AURKB (A) or Survivin (B) in PDX-expanded B-ALL blasts. (C-D) Quantification of the AURKB (C) and Survivin (D) fluorescence signal at the inner centromere; n = 30 metaphases from 3 non-HyperD and n = 30 metaphases from 3 HyperD-ALL. (E) Representative IF showing either centromeric and scattered localization of AURKB, Survivin, and CENP-A. (F-G) Frequency of PDX-expanded non-HyperD (n = 3) and HyperD-ALL (n = 3) blasts showing centromeric vs scattered localization of AURKB (n = 797 chromosomes from non-HyperD and n = 676 chromosomes from HyperD) (F), and Survivin (n = 964 chromosomes from non-HyperD and n = 814 chromosomes from HyperD) (G). (H) Quantification of total AURKB fluorescence signal from samples in panel C. AURKB levels are expressed relative to non-HyperD blasts, which are arbitrarily set to 100. (I) Left, representative FACS staining of H3S10P. Right, MFI of H3S10P in 3 non-HyperD and 3 HyperD-ALL samples. (J) Left, representative images of normal or railroad-shaped chromosomes. Right, frequency of metaphases showing the indicated number of chromosomes with PCS; n = 200 metaphases from 4 non-HyperD, n = 250 metaphases from 5 HyperD-ALLs samples. (K) Schematic depicting the workflow for functional analysis of the SAC. (L) Representative FACS of mitotic PDX-expanded B-ALL blasts (H3S10P+CD19+ cells, green) in the presence or absence of nocodazole. (M) Fold-change of mitotic (H3S10P+) blasts in nocodazol-treated (relative to DMSO-treated) non-HyperD (n = 3) and HyperD-ALL (n = 3) primary blasts. (N) Representative FACS cell cycle distribution of nocodazol- vs DMSO-treated PDX-expanded B-ALL blasts. (O) Quantification of the cell cycle phases in nocodazol- vs DMSO-treated PDX-expanded B-ALL blasts; n = 3 non-HyperD and n = 3 HyperD-ALL. (P) Quantitative reverse transcription–PCR analysis of SAC proteins in B-ALL primary samples; n = 9 non-HyperD and n = 11 HyperD. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .001; **** P < .0001 (2-way ANOVA or Student t test). Scale bars, 10 µm.
Mislocalized AURKB and Survivin from the inner centromere and loss of chromatid cohesion and SAC impairment in HyperD-ALL blasts. (A-B) Representative IF staining for CENP-A and AURKB (A) or Survivin (B) in PDX-expanded B-ALL blasts. (C-D) Quantification of the AURKB (C) and Survivin (D) fluorescence signal at the inner centromere; n = 30 metaphases from 3 non-HyperD and n = 30 metaphases from 3 HyperD-ALL. (E) Representative IF showing either centromeric and scattered localization of AURKB, Survivin, and CENP-A. (F-G) Frequency of PDX-expanded non-HyperD (n = 3) and HyperD-ALL (n = 3) blasts showing centromeric vs scattered localization of AURKB (n = 797 chromosomes from non-HyperD and n = 676 chromosomes from HyperD) (F), and Survivin (n = 964 chromosomes from non-HyperD and n = 814 chromosomes from HyperD) (G). (H) Quantification of total AURKB fluorescence signal from samples in panel C. AURKB levels are expressed relative to non-HyperD blasts, which are arbitrarily set to 100. (I) Left, representative FACS staining of H3S10P. Right, MFI of H3S10P in 3 non-HyperD and 3 HyperD-ALL samples. (J) Left, representative images of normal or railroad-shaped chromosomes. Right, frequency of metaphases showing the indicated number of chromosomes with PCS; n = 200 metaphases from 4 non-HyperD, n = 250 metaphases from 5 HyperD-ALLs samples. (K) Schematic depicting the workflow for functional analysis of the SAC. (L) Representative FACS of mitotic PDX-expanded B-ALL blasts (H3S10P+CD19+ cells, green) in the presence or absence of nocodazole. (M) Fold-change of mitotic (H3S10P+) blasts in nocodazol-treated (relative to DMSO-treated) non-HyperD (n = 3) and HyperD-ALL (n = 3) primary blasts. (N) Representative FACS cell cycle distribution of nocodazol- vs DMSO-treated PDX-expanded B-ALL blasts. (O) Quantification of the cell cycle phases in nocodazol- vs DMSO-treated PDX-expanded B-ALL blasts; n = 3 non-HyperD and n = 3 HyperD-ALL. (P) Quantitative reverse transcription–PCR analysis of SAC proteins in B-ALL primary samples; n = 9 non-HyperD and n = 11 HyperD. Graphs represent the mean, and error bars represent the standard error of the mean. *P < .05, **P < .01; ***P < .001; **** P < .0001 (2-way ANOVA or Student t test). Scale bars, 10 µm.
Defective AURKB is associated with loss of chromatid cohesion and SAC impairment in HyperD-ALL blasts
The confined localization of AURKB at the inner centromere is essential for chromatid cohesion and proper SAC activity.30,50,51 Indeed, analysis of chromosome spreads revealed that 75% of the metaphases from HyperD-ALL blasts displayed “railroad chromosomes,” a common phenotype reflecting premature chromatid separation (PCS) due to reduced chromatid cohesion at centromeres (Figure 5J; supplemental Figure 8a). These results were reproduced using B-ALL cell lines (supplemental Figure 8b).
AURKB controls chromosome biorientation/alignment at the metaphase plate through phosphorylation of different SAC proteins.30,50,52,53 We thus reasoned that AURKB defects may underlie the progression toward late mitosis of HyperD-ALL blasts with misaligned chromosomes by preventing SAC activation. To test this, we cocultured B-ALL primografts on Nestin-positive FBM-MSCs in the presence of nocodazol, which generates persistently unattached kinetochores leading to SAC activation and mitotic accumulation (Figure 5K), and found that nocodazol-treated HyperD-ALL blasts did not accumulate in mitosis as efficiently as non-HyperD-ALL blasts (Figure 5L-M). These results were reproduced in B-ALL cell lines (supplemental Figure 8c). Cell cycle analysis further confirmed that HyperD-ALL blasts do not arrest in G2/M after nocodazol treatment but they accumulate in G0/G1 (Figure 5N-O), strongly suggesting mitotic slippage. Apoptosis was not different between HyperD-ALL and non-HyperD-ALL blasts (supplemental Figure 8d). It is noteworthy that the metaphase-to-anaphase promoting regulator MAD2L2, whose loss leads to accelerated mitosis and mitotic aberrations,54 was found downregulated in HyperD-ALL blasts (Figure 5P). Collectively, HyperD-ALL cells show chromatid cohesion defects and undergo mitotic slippage most likely due to defective AURKB and impaired SAC activity, downstream of condensin-complex defects.
Inhibition of AURKB and SAC in CD34+ HSPCs reproduces chromosome-structure defects and hyperdiploid karyotypes
We next prompted to functionally model whether defective AURKB and impaired SAC activity could reproduce the phenotype observed in HyperD-ALL primary blasts. Because HyperD-ALL was shown to have a prenatal origin and preleukemic hyperdiploid precursors are found at birth,55,56 we exposed fetal BM-derived CD34+ HSPCs to the AURKB inhibitor ZM447439 and/or to the SAC inhibitor Reversine for 48 hours, and then processed cells for cytogenetics analysis (Figure 6A-B). Both AURKB and SAC inhibition in CD34+ HSPCs reproduced chromosome-structure defects observed in HyperD-ALL (Figure 6B; supplemental Figure 9a). They both drastically increased (compared with controls) the frequency of CD34+ HSPCs with micronuclei, a bona fide marker of chromosome instability (Figure 6C), hypocondensed chromosomes (Figure 6D), and metaphases with PCS reflecting loss of chromatid cohesion (Figure 6E). Such chromosome-structure defects were maintained and/or slighted potentiated when both AURKB and SAC were simultaneously inhibited (Figure 6B-E). Of note, although we could not reliably assess the karyotypes of reversine-treated CD34+ HSPCs due to massive chromosome damage, AURKB inhibition resulted in ∼30% of the CD34+ HSPCs displaying hyperdiploid karyotypes (Figure 6F; supplemental Figure 9b-c). In addition, cell cycle analysis revealed massive alterations in DNA ploidy, confirming genomic imbalances in CD34+ cells upon treatment with AURKB or SAC inhibitors (Figure 6G). The chromosome-structure defects and hyperdiploid karyotypes reproduced in CD34+ cells reinforce defective AURKB and SAC as an underlying cellular/molecular mechanism in hyperdiploidy B-ALL.
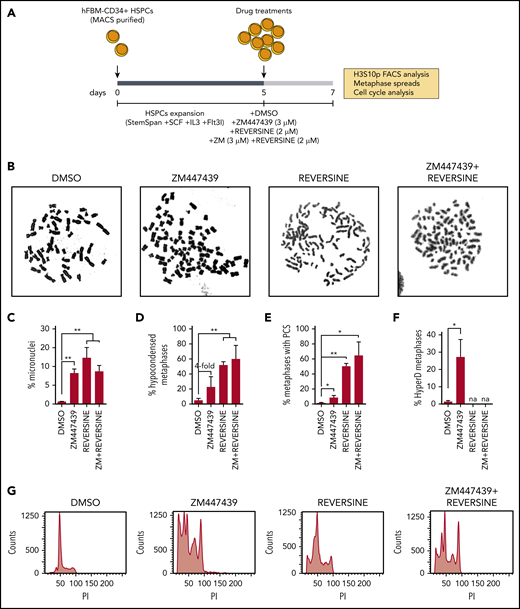
Inhibition of AURKB and SAC in CD34+ HSPCs reproduces chromosome structure defects and hyperdiploid karyotypes. (A) Schematic depicting the workflow for AURKB and SAC inhibition in CD34+ HSPCs. (B) Representative images of metaphase chromosomes treated as indicated. (C-F) Frequency of metaphases with micronuclei (n = 500 cells per experiment) (C), hypocondensed chromosomes (D), PCS (E), and hyperdiploidy karyotype (F). (G) Representative FACS analysis showing PI staining profiles in CD34+ HSPCs treated as indicated; n = 150 metaphases analyzed per treatment from 3 independent experiments. *P < .05, **P < .01 (1-tailed Student t test).
Inhibition of AURKB and SAC in CD34+ HSPCs reproduces chromosome structure defects and hyperdiploid karyotypes. (A) Schematic depicting the workflow for AURKB and SAC inhibition in CD34+ HSPCs. (B) Representative images of metaphase chromosomes treated as indicated. (C-F) Frequency of metaphases with micronuclei (n = 500 cells per experiment) (C), hypocondensed chromosomes (D), PCS (E), and hyperdiploidy karyotype (F). (G) Representative FACS analysis showing PI staining profiles in CD34+ HSPCs treated as indicated; n = 150 metaphases analyzed per treatment from 3 independent experiments. *P < .05, **P < .01 (1-tailed Student t test).
Discussion
This is the most comprehensive study to date on the cellular mechanisms underlying the mitotic and chromosome defects contributing to the pathophysiology of pediatric HyperD-ALL. Here, we have developed robust in vitro assays using Nestin-positive fetal BM-MSCs and in vivo PDX to successfully expand primary B-ALL leukemic samples. Such ex vivo and in vivo expansion of B-ALL leukemic samples provided enough mitotic/dividing primary blasts to address many biological questions in a large cohort of 54 primary B-ALL samples, thus highlighting the clinical relevance of our work. The only available HyperD-ALL cell line, CALL-2, was used throughout the study for confirmatory and gain-of-function studies, and consistently phenocopied the data generated using primary HyperD-ALL primary cells. This cell line originates from a doubled-up hypodiploid B-ALL,23 suggesting that the same mitotic/chromosomal defects here reported may underlie the pathogenesis of hypodiploid B-ALL patients. Future studies, however, should be done in HypoD-ALL patients who are rare but clinically dismal.
We demonstrate that, in contrast to nonHyperD-ALL, HyperD-ALL blasts show reduced proliferative rates coupled with a delay in early mitosis at prometaphase. Such a delay in early mitosis is associated with chromosome-alignment defects at the metaphase plate, which, in fact, lead to chromosome-segregation defects and nonmodal karyotypes. Despite karyotype heterogeneity, HyperD-ALL primary blasts show a major clone that most likely represents the fittest clone after cell adaptation to aneuploidy.57 These data support previous studies showing the presence of cytogenetically different subclones in HyperD-ALL.58-61
Mechanistically, HyperD-ALL primary blasts and cell lines did not show abnormalities in bipolar spindle and kinetochore formation. However, they displayed important chromosome-structure and -function defects, a major mechanism regulating chromosome segregation that is essential for maintaining the genomic integrity of cells.13-15 HyperD-ALL cells showed robust defects in several members of the condensin complexes including SMC2 (levels reduced at chromosome scaffolds), CAPD2, and CAPD3. Biochemical, functional, and MS assays revealed that PTMs of condensin-complex proteins may represent a mechanism underlying defective condensin complexes in HyperD-ALL cells. Of note, no mutations were found in condensin complex encoding genes in HyperD-ALL patients, ruling out genomic mutations as the cause of the defective condensin complex in HyperD-ALL patients.
Consequently, high-order chromosome-architecture defects are notorious, and include chromosome hypocondensation and loss of centromere rigidity revealed by increased intercentromeric distances. Consistently, a recent study has reported chromosome architecture defects and lower expression of CTCF in HyperD-ALL samples.62 Furthermore, AURKB, the catalytic subunit of the CPC, and Survivin are mislocalized from the inner centromere in early mitosis, further linking defective condensin with chromosome-segregation defects in HyperD-ALL blasts. According to the essential localization of AURKB at the inner centromere to protect chromatid cohesion and for proper SAC activity,30,50,51 HyperD blasts show chromatid-cohesion defects as observed by PCS and impaired SAC, leading to mitotic slippage. Impaired SAC activity explains why HyperD-ALL blasts proceed to late mitosis with misaligned chromosomes at the metaphase plate, thus leading to chromosome-segregation defects. Importantly, chromosome hypocondensation and hyperdiploidy were functionally reproduced in CD34+ HSPCs upon inhibition of AURKB and/or SAC, reinforcing defective condensin complex, AURKB, and SAC as underlying cellular and molecular mechanisms in HyperD-ALL (Figure 7). Although they are likely instrumental in the pathophysiology of the HyperD-ALL, whether these findings are causal or consequential to hyperdiploidy remains an open question under investigation.

Proposed model of the cellular mechanisms underlying the mitotic and chromosome defects contributing to the pathophysiology of pediatric HyperD-ALL. (A) Non–HyperD-ALL. (B) HyperD-ALL.
Proposed model of the cellular mechanisms underlying the mitotic and chromosome defects contributing to the pathophysiology of pediatric HyperD-ALL. (A) Non–HyperD-ALL. (B) HyperD-ALL.
This is the first cellular and molecular in-depth characterization of the mitotic and chromosomal defects of HyperD-ALL using a cohort of 54 B-ALL primary samples. It represents a highly relevant study because B-ALL is the most common pediatric cancer. Studies in monozygotic twins with concordant HyperD-ALL and retrospective analysis of HyperD clones in cord blood indicated that HyperD clones arise prenatally,7 and that hyperdiploidy is an initiating oncogenic event generating a preleukemic clone that then requires secondary mutations to trigger a full-blown leukemia.63 Therefore, a better mechanistic understanding of how hyperdiploidy occurs and how secondary alterations are acquired becomes crucial not only to propose novel therapeutic targets but also to prevent the progression/relapse of HyperD-ALL. These defects in condensin complex-AURBK-SAC axis open up new avenues for modeling HyperD-ALL by genetically engineering HSPCs, which will be crucial to further address the causal contribution of these defects to the origin of HyperD in B-ALL.
From a diagnostic-clinical standpoint, the high-order chromatin/chromosome structural defects observed in HyperD-ALL explain very well the difficulties that clinical cytogeneticists have historically encountered to obtain metaphases of standard quality from these patients, thus challenging the cytogenetic diagnostic.3 In addition, despite the favorable clinical outcome of HyperD-ALL, unraveling the physiopathogenic mechanisms underlying HyperD-ALL is necessary because in absolute numbers, the morbidity/mortality associated with HyperD-ALL still represent a clinical challenge. In sum, this study sheds light on the mechanisms underlying the mitotic and chromosome defects involved in the pathogenesis of HyperD-ALL and offers molecular targets (condensin-complex members, CPC members, AURKB, or the SAC) for potential pharmacological intervention in the most frequent molecular subtype of pediatric acute leukemia.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors dedicate this paper to the memory of their collaborator Anna Bosch (Scientific and Technological Centers, Universitat de Barcelona [CCiTUB], Barcelona, Spain) and to their patients. The authors were particularly inspired by a patient with hyperdiploid B-ALL who died after relapse. The authors thank Dolors Costa (Hospital Clínic, Barcelona, Spain), Elisenda Coll (CCiTUB, Barcelona, Spain); Samanta Zanneti, Talía Velasco, Raúl Torres, and Belén López-Millán (laboratory of P.M.) for technical input; Ana Losada (Centro Nacional de Investigaciones Oncológicas [CNIO], Madrid, Spain) for providing condensin Ab; M. Alba Abad and William C. Earnshaw (University of Edinburgh, Edinburgh, United Kingdom) for critical discussions; and Anthony Moorman (University of Newcastle, Newcastle upon Tyne, United Kingdom) and Anindita Roy (Oxford University) for providing whole-genome and RNA-seq data, respectively. The authors thank “Biobanc de l’Hospital Infantil Sant Joan de Déu per la Investigació,” integrated into the Spanish Biobank Network of ISCIII, for sample and data procurement. The authors thank Centres de Recerca de Catalunya (CERCA)/Generalitat de Catalunya and Fundació Josep Carreras‐Obra Social la Caixa for their institutional support.
This work was supported by the European Research Council (CoG-2014-646903 [P.M.]), the Spanish Ministry of Economy and Competitiveness (SAF-2016-80481-R [P.M.]), the Asociación Española Contra el Cáncer (AECC-CI-2015), and the ISCIII (PI17/01028) (C.B.). The authors also acknowledge support from Fundación Leo Messi and the Fundación Uno entre Cienmil. O.M. was supported by a Lady Tata Memorial Trust (2017) and a “Beatriu de Pinós” postdoctoral fellowship (2018-2019) from Generalitat de Catalunya. D.P. was supported by an NC3Rs fellowship and a Children’s Cancer and Leukemia Group (CCLG) project grant. This work was also supported by the Spanish Ministry of Economy and Competitiveness (SAF-2016-75286-R [R.R.]), the ISCIII/FEDER (Miguel Servet Program CPII16/00049 [R.R.], Sara Borrell Program CD16/00103 [S.T.M.], and PI16/00280), and Consorcio CIBERONC (CB16/12/00390).
P.M. is an investigator of the Spanish Cell Therapy Network (TERCEL).
Authorship
Contribution: O.M. conceived the study, designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript; C.B. designed and performed experiments and analyzed and interpreted the data; M.V., L.V., C.M.L.-L., P.R.-G., I.G., H.R.-H., F.G.-A., J.L.T., S.T.M., D.P., R.R., and M. Calvo performed experiments and analyzed data; P.B., M.L.d.B., I.P., M.M.P.-I., S.R.-P., M.-J.C., M.R.-O., and M. Camós provided patient samples and data; and P.M. conceived the study, designed experiments, interpreted the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Oscar Molina, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Casanova 143, 08036, Barcelona, Spain; e-mail: omolina@carrerasresearch.org; or Pablo Menendez, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Casanova 143, 08036, Barcelona, Spain; e-mail: pmenendez@carrerasresearch.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal