


Abstract
Avoiding immune destruction is a hallmark of cancer. Over the past few years, significant advances have been made in understanding immune dysfunction and immunosuppression in multiple myeloma (MM), and various immunotherapeutic approaches have delivered improved clinical responses. However, it is still challenging to completely eliminate malignant plasma cells (PCs) and achieve complete cure. The interplay between the immune system and malignant PCs is implicated throughout all stages of PC dyscrasias, including asymptomatic states called monoclonal gammopathy of undetermined significance and smoldering myeloma. Although the immune system effectively eliminates malignant PCs, or at least induces functional dormancy at early stages, malignant PCs eventually evade immune elimination, leading to progression to active MM, in which dysfunctional effector lymphocytes, tumor-educated immunosuppressive cells, and soluble mediators coordinately act as a barrier for antimyeloma immunity. An in-depth understanding of this dynamic process, called cancer immunoediting, will provide important insights into the immunopathology of PC dyscrasias and MM immunotherapy. Moreover, a growing body of evidence suggests that, together with nonhematopoietic stromal cells, bone marrow (BM) immune cells with unique functions support the survival of normal and malignant PCs in the BM niche, highlighting the diverse roles of immune cells beyond antimyeloma immunity. Together, the immune system critically acts as a rheostat that fine-tunes the balance between dormancy and disease progression in PC dyscrasias.
Introduction
Multiple myeloma (MM) is a plasma cell (PC) dyscrasia that is characterized by the uncontrolled proliferation of malignant PCs within the bone marrow (BM), as well as clinical symptoms, such as bone destruction, kidney injury, and paraproteinemia.1 The clinical spectrum of PC dyscrasias also includes a benign expansion of PCs, called monoclonal gammopathy of undetermined significance (MGUS), and a more advanced premalignant condition known as smoldering MM (SMM).2 Despite great therapeutic progress over the last decade, active MM remains an incurable disease as a result of clonal heterogeneity of malignant PCs that is responsible for recurrent relapses with shorter periods of remissions. Genetic alterations within malignant PCs play a critical role in MM clinical outcomes.3 Besides the well-known del(17p) and t(4;14), several additional cytogenetic factors are involved in active MM evolution, such as del(1p32), gain(1q), and trisomies.4 Interestingly, these alterations are often present in transformed PC clones from MGUS and SMM stages,5 suggesting that extrinsic factors govern the balance between latency (ie, dormancy) and the progression of preneoplastic lesions. In this context, the immune system has a strong impact on this balance. This review highlights the dynamic cross talk between tumor cells and the immune system in PC dyscrasias, which provides important information in the era of immunotherapy.
Immune cells as cellular components supporting PC survival
The BM niche is the primary residence of long-lived PCs, and the survival of normal and malignant PCs is supported by the complex interplay among cellular components, extracellular matrix (ECM) proteins, and soluble factors.6 Historically, mesenchymal stem cells (MSCs) have been recognized as key players by their multifaceted roles, including homing of PCs in the BM by the secretion of CXCL12 (the ligand of CXCR4), contact-dependent support of PCs by integrins, and secretion of prosurvival factors, such as interleukin-6 (IL-6) and vascular endothelial growth factor (VEGF). The reciprocal interactions between MM cells and MSCs play an important role in MM pathogenesis.7 Indeed, MM-educated MSCs acquire the ability to produce high amounts of proinflammatory cytokines and growth factors that favor accumulation and chemoresistance of malignant PCs.8,9
In contrast to the hematopoietic stem cell (HSC) niche, the exact localization of PCs in the BM niche remains to be fully characterized.10 Under chronic inflammatory conditions, the accumulation of PCs is also observed in inflamed tissues, raising a possibility that multiple cellular components might be able to support the survival of PCs in a redundant or compensatory manner.10 In this context, immune cell subsets, especially myeloid-lineage cells, have been recognized as important players for survival and maintenance of PCs in the BM. Eosinophils can produce IL-6 and APRIL, and the numbers of BM PCs and mucosal IgA+ PCs were reduced in mice deficient for eosinophils.11,12 Although it remains controversial whether eosinophils are indispensable for the normal BM PC pool,13,14 these cells have the ability to stimulate the proliferation of malignant PCs. Wong et al showed that eosinophils were located in close proximity to malignant PCs in BM biopsy samples, and eosinophil-derived soluble factors stimulated the proliferation of malignant PCs cocultured with MSCs in a nonredundant manner.15 More recently, by analyzing gut microbiota in the Vk*MYC transgenic mice developing de novo MM, Calcinotto et al showed that a commensal bacteria, Prevotella heparinolytica, promoted the differentiation of T helper 17 (Th17) cells and that IL-17–driven activation of BM eosinophils supported MM progression.16 Tumor-associated dysbiosis and its impact in patients with PCs will require further investigation.
Beyond their antigen-presentation capacity, CD11c+ conventional dendritic cells (cDCs) that reside in the BM play an important role in the biology of PCs and may also be involved in MM pathogenesis. Indeed, the interaction between CD28 on PCs and CD80/CD86 on cDCs was shown to promote the survival of PCs, either by triggering dendritic cell (DC)-derived IL-6 release17 or by activating CD28-mediated downstream prosurvival signaling.18 CD28 signaling is negatively regulated by CTLA-4, as a result of its higher avidity for CD80/CD86. Interestingly, a recent study showed that CTLA-4+ BM regulatory T cells (Tregs) are colocalized with PCs and CD11c+ cDCs, as well as that BM Tregs support the maintenance of long-lived PCs, possibly by limiting PC activity.19 This cross talk among Tregs, cDCs, and malignant PCs might be relevant for MM cell dynamics within the BM. Plasmacytoid DCs (pDCs) are another major DC subset that is predominantly derived from a lymphoid progenitor. Like cDCs, pDCs were shown to accumulate in the active MM BM and to promote growth, survival, chemotaxis, and chemotherapy resistance in a contact-dependent manner.20 Additionally, pDCs have the ability to produce high levels of type 1 interferon (IFN) and IL-6,21 both of which may fuel MM growth.
As observed in solid malignancies, MM progression is tightly associated with dynamic alteration of the surrounding environment, triggering angiogenesis and ECM remodeling in the BM niche. Above all, osteolytic bone disease is a key hallmark of MM-associated pathology. The interaction between MM and MSCs induces secretion of pro-osteoclastogenic factors, including proinflammatory cytokines and RANKL, leading to activation of osteoclasts and suppression of osteoblasts.22 In the 5TGM1 preclinical MM model, adoptively transferred CD11b+Gr-1+ immature myeloid cells were shown to differentiate into osteoclasts.23 Although these results suggest that the MM niche favors the generation of osteoclasts from monocytic cells, BM-resident macrophages might also support MM cells, given that depletion of CD169+ macrophages by clodronate liposomes remarkably inhibited the engraftment of 5TGM1 MM cells.24 Because BM-resident macrophages retain HSCs,25,26 certain subsets of macrophages might be involved in contact-dependent maintenance of MM cells.
Notably, accumulation of normal PCs themselves may have a strong impact on the BM microenvironment. Pioli et al recently showed that PCs accumulate in the aged BM and that depletion of PCs attenuated age-associated and myeloid-skewed hematopoiesis.27 Mechanistically, aged PCs had high expression of genes related to Toll-like receptor (TLR) signaling, and their interaction with MSCs resulted in inflammation and myelopoiesis.27 Thus, the link between aged PCs and the proinflammatory BM milieu could provide important insights into the immunopathology of PC dyscrasias; further studies are warranted.
Together, a variety of immune cells support the normal and malignant PCs as cellular components of the BM niche. However, little is known regarding the temporal and spatial distribution patterns of BM immune subsets clustering with malignant PCs. These immune cell subsets supporting MM cells might be involved in tumor dormancy and reactivation, which will be discussed in the next section.
Dormancy and cancer immunoediting in MM
Cancer dormancy is a heterogeneous phenomenon that has been defined differently, depending on the context.28,29 Cellular dormancy is a long-term quiescent state that can be observed at the level of the single tumor cell. By contrast, more advanced tumors can also undergo functional dormancy (ie, tumor mass dormancy), in which the balance between tumor proliferation and tumor regression is regulated by extrinsic factors (Figure 1).
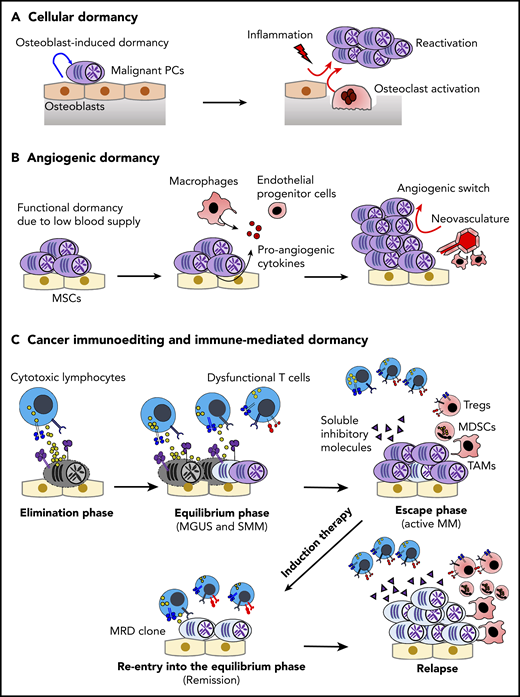
Dormancy and cancer immunoediting in PC dyscrasias. (A) The BM osteoblastic niche induces dormancy in malignant PCs. Note that other immune cells in the PC niche might also contribute to cellular dormancy. Enhanced osteoclast activity and inflammation signaling can abrogate the niche-induced dormancy. (B) Lack of sufficient blood supply renders functional dormancy, called angiogenic dormancy. In addition to the bidirectional interaction between MM cells and MSCs, tumor-associated macrophages (TAMs) contribute to the generation of proangiogenic factors. In response to these stimuli, endothelial progenitor cells and TAMs cooperatively generate neovasculature, which might trigger the growth of MM. (C) Cytotoxic lymphocytes, such as natural killer cells and CD8+ T cells, critically contribute to the immunosurveillance of malignantly transformed cells in the elimination phase. In the equilibrium phase, immune-mediated functional dormancy prevents outgrowth of malignant PCs, which might represent MGUS and SMM. In active MM, malignant PCs eventually overwhelm the immune system, leading to development of the immunosuppressive milieu (the escape phase). Reentry into the equilibrium phase can be achieved by remission-induction therapy, which can contribute to long-term minimal residual disease (MRD) control in some patients. However, the immunosuppressive milieu is reestablished after relapse.
Dormancy and cancer immunoediting in PC dyscrasias. (A) The BM osteoblastic niche induces dormancy in malignant PCs. Note that other immune cells in the PC niche might also contribute to cellular dormancy. Enhanced osteoclast activity and inflammation signaling can abrogate the niche-induced dormancy. (B) Lack of sufficient blood supply renders functional dormancy, called angiogenic dormancy. In addition to the bidirectional interaction between MM cells and MSCs, tumor-associated macrophages (TAMs) contribute to the generation of proangiogenic factors. In response to these stimuli, endothelial progenitor cells and TAMs cooperatively generate neovasculature, which might trigger the growth of MM. (C) Cytotoxic lymphocytes, such as natural killer cells and CD8+ T cells, critically contribute to the immunosurveillance of malignantly transformed cells in the elimination phase. In the equilibrium phase, immune-mediated functional dormancy prevents outgrowth of malignant PCs, which might represent MGUS and SMM. In active MM, malignant PCs eventually overwhelm the immune system, leading to development of the immunosuppressive milieu (the escape phase). Reentry into the equilibrium phase can be achieved by remission-induction therapy, which can contribute to long-term minimal residual disease (MRD) control in some patients. However, the immunosuppressive milieu is reestablished after relapse.
Cellular dormancy in MM
As the primary site for hematopoiesis, the BM niche critically contributes to the long-term maintenance of HSCs by inducing quiescence under steady conditions.30 Several pieces of evidence suggest that tumor cells disseminated in the BM can hijack the BM niche, allowing them to enter into dormancy.29 For example, although early metastatic BM foci are often detected in cancer patients, not all patients develop metastatic bone disease, which often occurs after a long latency period (>10 years after surgical resection of the primary tumor).29 In this context, Chen et al first showed that MM cells in the osteoblastic niche possess cellular quiescence or stem-like features.31 It is appreciated that normal HSCs are localized in relatively close proximity to the perivascular niche, whereas lymphoid progenitors seem to reside preferentially in the osteoblastic niche,32,33 suggesting a differential requirement of the BM niche for normal HSCs and MM cells. The critical role of the osteoblastic niche for MM dormancy was further demonstrated by intravital imaging–based tracking of the 5TGM1 MM model.34 Although osteoblasts conferred dormancy and chemoresistance to MM cells, the RANKL-driven activation of osteoclasts was shown to disrupt the osteoblastic niche–induced dormancy,34 highlighting the dynamic plasticity of dormancy and reactivation in the BM microenvironment. This result might explain why patients treated with antiosteoclastic agents, such as denosumab35,36 or zoledronic acid,37 showed improved progression-free survival. Currently, denosumab is being tested in SMM patients (NCT03839459), and these results may provide further evidence of the importance of osteoclasts in MM development. In addition, recent molecular mechanisms regulating the balance between dormant and active MM cells were identified by single-cell RNA sequencing.38 Intriguingly, several genes encoding transcriptional factors and receptors related to myeloid-lineage cells were upregulated in the niche-induced dormant MM cells. Among dormancy-related myeloid signature genes, AXL (a member of the tumor-associated macrophage [TAM] family of receptor tyrosine kinases) may play a predominant role, given that inhibition of AXL by small molecule inhibitors triggered the reactivation of dormant MM cells. In addition, the expression levels of AXL and coregulated genes were higher in PCs from MGUS compared with MM, and higher expression level of the dormant myeloid signature genes was associated with better prognosis in patients with active MM.38 Still, the functional roles of these myeloid-related genes in dormant MM cells are yet to be characterized. One possibility is that inflammatory stimuli might also trigger the reactivation of MM cells with a dormant myeloid transcriptome signature. Indeed, Kikuchi et al showed that lipopolysaccharide stimulation can reactivate dormant MM cells via CD180/MD-1 noncanonical lipopolysaccharide receptor expressed on MM cells, leading to accelerated disease progression.39 Thus, it is possible that infection and inflammation might abrogate cellular dormancy in patients with PC dyscrasias, either by enhancing osteoclast activities or by direct inflammatory signaling in malignant PCs. Further studies are warranted to understand crucial cellular and molecular events triggering the reactivation of dormant tumor cells.
Angiogenic dormancy
The availability of oxygen and nutrients via new vasculature is critical for tumor growth. Although angiogenic dormancy in MM is yet to be proven, substantial evidence supports that angiogenesis is a key driver for MM progression. Indeed, there is a positive correlation between the levels of BM microvessel density and disease stages in active MM.40 Additionally, the frequency of circulating endothelial progenitor cells is significantly higher in patients with active MM compared with healthy subjects,41 suggesting that these cells might be mobilized into the MM niche to generate neovasculature. The cross talk between MM cells and MSCs that favors the secretion of VEGF may play an important role in MM neoangiogenesis. In addition, macrophages might represent another key player through their secretion of proangiogenic factors and their contact-dependent microvascular remodeling.42 Their importance is supported by a positive correlation between the infiltration of CD163+ M2-like TAMs and high-grade microvessel density in MM.43 Another notable feature of MM TAMs is vasculogenic mimicry. Scavelli et al showed that BM macrophages isolated from patients with active MM, but not from normal subjects or MGUS patients, can organize capillary-like structures in response to VEGF,44 indicating that MM TAMs have been reprogrammed to promote angiogenesis. Of note, in other malignancies, various types of immune cells (eg, neutrophils, Tregs, and natural killer [NK] cells) are also reprogrammed to secrete proangiogenic factors.45-47 Moreover, cell type–specific deletion of VEGF in cytotoxic lymphocytes48 or NK cells49 dramatically alters angiogenesis and tumor growth, highlighting the critical role of immune-mediated angiogenesis. Thus, multiple immune cells could be involved in the angiogenic switch associated with MM development; future studies will have to be performed to understand their relative roles.
Immune-mediated dormancy and cancer immunoediting
Immune-mediated dormancy is considered a part of the cancer immunoediting process, which is composed of 3 phases: elimination, equilibrium (ie, immune-mediated dormancy), and escape.50 In the elimination phase, innate and adaptive immunity continuously recognize and eliminate malignant transformed cells, and cellular homeostasis is maintained by immune surveillance.50 Because clinically detectable tumors have already established tumor mass as a consequence of evasion from immune surveillance, essentially all malignancies at diagnosis represent the escape phase. However, significant evidence indicates the existence of a dynamic equilibrium phase between the elimination phase and the escape phase.51-53 In the equilibrium phase, immune-mediated functional tumor dormancy can contribute to long-term disease latency, although tumors ultimately overwhelm the immune system by clonal evolution of tumor cells under immune selective pressure and/or by alteration of the immune microenvironment.50,54
Cancer immunoediting has only been partly characterized in PC dyscrasias. Still, as shown in other malignant pathologies, accumulating experimental evidence supports the critical roles of NK cells and CD8+ T cells and their effector molecules perforin and IFN-γ in myeloma recognition and elimination. CD226 (DNAM-1), an adhesion receptor that controls NK and CD8+ T cell activation, seems to play a major role in MM elimination.55-57 Indeed, its ligands CD112 and CD155 are often overexpressed by myeloma cells as a consequence of cellular stress,57 and, in the de novo Vk*MYC myelomagenesis model, CD226 deficiency accelerated paraproteinemia and shortened survival.55 Also, another well-characterized receptor, receptor group 2, member D (NKG2D), which binds to stress-induced ligands major histocompatibility complex class I–related chains A and B (MICA/B), and UL16 binding proteins may also be involved in the elimination of MM by NK cells and CD8+ T cells. As a consequence, tumor-derived soluble NKG2D ligands (eg, soluble MICA) were shown to dampen NKG2D functions by ligand-induced receptor downregulation and were suggested to promote MM immune escape.58 Intriguingly, MGUS patients, but not patients with active MM, exhibit high-titer anti-MICA antibodies that antagonize the inhibitory effect of soluble MICA,59 suggesting that humoral immunity and NKG2D-mediated immunosurveillance might cooperatively control disease progression. However, it remains largely unknown whether dysfunctional humoral immunity, a key feature in PC dyscrasias, has a direct impact on immunosurveillance.
Like other malignancies, the equilibrium phase has not been stringently demonstrated because of technical difficulties, but asymptomatic PC dyscrasias (MGUS and SMM) might represent this phase.2 Indeed, xenografted PCs from MGUS patients show progressive growth in immunodeficient humanized mice,60 suggesting the involvement of extrinsic control in the disease latency. Notably, antigen-specific T-cell responses against premalignant PCs are reported in patients with MGUS, and several antigens have been identified, such as SOX2 (an embryonic stem cell antigen) and OFD1.61-63 Dhodapkar et al performed a prospective analysis of antigen-specific T cells against SOX2 in 305 patients with MGUS or asymptomatic MM. They showed that the absence of anti-SOX2 T cells is significantly associated with future progression to active MM,64 highlighting the importance of tumor-specific T cells for preventing disease progression. In high-risk SMM patients, lenalidomide (an immune-modulating drug), as monotherapy65 or in combination with low-dose dexamethasone,66 significantly delayed progression to the active MM stage. Additionally, functionally restored NK cells and CD8 T cells were observed in the treated group,67 suggesting that immune-modulating drugs may improve immune-mediated dormancy.
Key factors triggering disease progression from the equilibrium phase to the escape phase remain largely unknown. Obviously, selection of immune-resistant clones might be responsible for immune escape. Additionally, in solid malignancies, tumor-specific T-cell–derived IFN-γ is known to trigger copy number alterations associated with DNA damage responses,68 raising the possibility that genetic instability induced by immune cells might be a driver for disease progression. Although the impact of immunity on clonal evolution and MM progression remains to be elucidated, significant progress has been made in understanding immune dysfunction and immunosuppression in active MM (as discussed in the next section). It should be noted that the transition from the equilibrium phase to the escape phase can be, to some degree, a reversible process in MM. A certain proportion of patients can achieve a complete response, with or without detectable minimal residual disease, using novel antimyeloma agents and autologous stem cell transplantation (ASCT). Indeed, using a preclinical model of BM transplantation (BMT), Vuckovic et al showed that donor-derived T cells and IFN-γ are indispensable for long-term disease control against transplantable Vk*MYC cells, whereas donor-derived IL-17 promotes the post-BMT relapse by directly inducing proliferation of residual MM cells.69 Again, lenalidomide-based post-ASCT maintenance therapy might also improve immune-mediated dormancy against MRD.
Together, the dynamic interplay among tumor cells, stromal cells, and immune cells orchestrates dormancy and cancer immunoediting in MM (Figure 1). Further studies are necessary to gain insights into the impacts of these processes on clonal selection and evolution of malignant PCs.
Immune dysfunction and immunosuppression in the MM niche
As discussed in the previous section, essentially all clinically diagnosed cancers have an established immunosuppressive milieu that allows tumor cells to evade antitumor immunity. The tumor microenvironment (TME) can be differentially organized depending on tumor genotype and phenotype, disease stage, treatment history, and patient background.54 However, the complex mechanisms of immunosuppression can be summarized as the interplay of 3 factors: dysfunction and/or exclusion of effector lymphocytes, generation and mobilization of immunosuppressive cells, and metabolites and cytokines that dampen antitumor immunity (Figure 2).
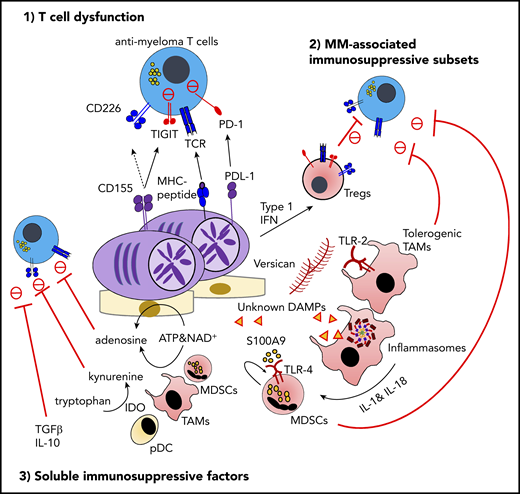
The immunosuppressive microenvironment in MM. TIGIT is frequently expressed on functionally exhausted T cells in the MM BM. At the interface between T cells and malignant PCs, the interaction between TIGIT and its ligand CD155 plays a critical role in negative regulation, including the competitive inhibition of CD226-dependent antitumor immunity. The proinflammatory MM milieu is critically implicated in the generation of immunosuppressive subsets, such as type 1 IFN–induced Tregs, versican-induced tolerogenic TAMs, and myeloid-derived suppressor cells (MDSCs) induced by S100A9 and the inflammasome-derived IL-18. Various soluble metabolites and cytokines derived from immunosuppressive subsets or malignant PCs also regulate effector lymphocyte functions. These factors include adenosine driven by ectoenzymes, indoleamine 2,3-dioxygenase (IDO)-induced tryptophan catabolites, transforming growth factor-β (TGF-β), and IL-10. ATP, adenosine triphosphate; DAMP, damage-associated molecular pattern; MDSC, myeloid-derived suppressor cell; MHC, major histocompatibility complex; NAD, nicotinamide adenine dinucleotide; PD-1, programmed cell death protein 1; PDL-1, programmed death ligand 1; TIGIT, T-cell immunoreceptor with immunoglobulin and ITIM domains.
The immunosuppressive microenvironment in MM. TIGIT is frequently expressed on functionally exhausted T cells in the MM BM. At the interface between T cells and malignant PCs, the interaction between TIGIT and its ligand CD155 plays a critical role in negative regulation, including the competitive inhibition of CD226-dependent antitumor immunity. The proinflammatory MM milieu is critically implicated in the generation of immunosuppressive subsets, such as type 1 IFN–induced Tregs, versican-induced tolerogenic TAMs, and myeloid-derived suppressor cells (MDSCs) induced by S100A9 and the inflammasome-derived IL-18. Various soluble metabolites and cytokines derived from immunosuppressive subsets or malignant PCs also regulate effector lymphocyte functions. These factors include adenosine driven by ectoenzymes, indoleamine 2,3-dioxygenase (IDO)-induced tryptophan catabolites, transforming growth factor-β (TGF-β), and IL-10. ATP, adenosine triphosphate; DAMP, damage-associated molecular pattern; MDSC, myeloid-derived suppressor cell; MHC, major histocompatibility complex; NAD, nicotinamide adenine dinucleotide; PD-1, programmed cell death protein 1; PDL-1, programmed death ligand 1; TIGIT, T-cell immunoreceptor with immunoglobulin and ITIM domains.
Dysfunction of effector lymphocytes in MM
MM-specific T-cell responses were first reported in the early 2000s.70,71 Most of the patients with active-stage MM expressed ≥1 cancer/testis antigen, such as MAGE, BAGE, GAGE, and LAGE-1/NY-ESO-172,73 ; indeed, oligoclonal cancer/testis antigen–specific CD8 T cells were detectable in a proportion of MM patients.74 Based on these results, significant efforts have been made to identify tumor-associated antigens and neoantigens that can be used for vaccine-based approaches.75,76 Perumal et al recently performed in silico analyses to determine MM neoantigens, and they identified several immunogenic neoantigens that elicit antimyeloma T-cell responses. Intriguingly, mutation and neoantigen burden seems to increase in relapsed MM patients compared with newly diagnosed MM patients,77 and high mutation and neoantigen loads were recently associated with shortened survival in newly diagnosed MM.78 This suggests that, although a higher tumor neoantigen load may provide more targets for T-cell–mediated disease control, the overall impact on clinical outcomes may be unfavorable. Alternatively, these results may be due to multiple confounding factors associated with high tumor mutation burden, such as chromosomal alterations that critically impact disease aggressiveness and clinical outcome.4 Additionally, the quality of neoantigen that elicits T-cell responses and potential T-cell dysfunction due to MM-intrinsic and/or environmental factors should be taken into account.
Altered polarization of T cells, particularly Th17-skewed immunity, has been reported in patients with active MM and might be driven by IL-1, IL-6, and transforming growth factor-β (TGF-β) in the proinflammatory MM niche.79 Importantly, IL-17 derived from Th17 cells can directly fuel tumor growth in MM, providing a possible therapeutic target.79,80 Recent comprehensive immune landscape analyses by single-cell RNA sequencing and mass cytometry revealed the substantial heterogeneity of T-cell subsets in MM,81-83 although the different functional roles of each subset remain unknown. It should be noted that these analyses might have limitations in MM as a result of its localization in the BM. Given that BM acts as a reservoir of memory T cells, as well as a primary lymphoid organ,84 tumor-specific T cells might be less abundant compared with the TME in solid malignancies. Also, hemodilution of BM aspirates remains a barrier to accurate analyses. Still, these studies suggest that MM progression might progressively sculpt unique and complex immune phenotypes in the BM, even in potential bystander immune cells. Notably, even at the MGUS stage, several key immune changes have already been initiated, including an increase in the number of terminal effector T cells and group 1 innate lymphoid cells.81,85 Although stem-like tissue-resident T cells are observed in MGUS patients, T cells from advanced MM patients are characterized by the loss of this subset and the emergence of senescent and exhausted T cells,81,86,87 suggesting that disease progression dynamically alters T-cell phenotypes.
Of note, although some evidence points toward an accumulation of hypoproliferative senescent CD57+CD8+ T cells in MM, the presence of exhausted T cells is controversial in newly diagnosed patients. Indeed, CD8+ T cells from newly diagnosed MM patients rarely express high levels of multiple immune checkpoint receptors (eg, PD-1, CTLA-4, TIM-3, and LAG-3), which represent one of the cardinal features of T-cell exhaustion.88 Recent phase 3 clinical trial results showed limited clinical benefits of anti–PD-1 blockade therapy in MM patients,89,90 also highlighting the importance of careful evaluation of the functional status of MM-specific T cells. In this context, TIGIT (T-cell immunoreceptor with immunoglobulin and ITIM domains), an inhibitory counterpart of the CD226 receptor, has emerged as a new immune checkpoint in MM.88,91 TIGIT has multiple inhibitory mechanisms, including competitive inhibition of CD226 at the cytotoxic synapse by sharing the same ligands, bidirectional negative regulation at the T- cell–DC interface, and inhibition by TIGIT+ Tregs.92 Notably, among immune checkpoint molecules, TIGIT is most frequently expressed on BM CD8 T cells from MM patients, and TIGIT+ T cells represent functionally exhausted phenotypes.88 TIGIT blockade can reinvigorate exhausted T cells, and it improved disease control in preclinical Vk*MYC models as a single agent88 or post-BMT immunotherapy.91 These results highlight the critical role of TIGIT in MM-specific T cells, but further studies are warranted to understand the molecular mechanisms regulating TIGIT expression and potential combination approaches.
Therapy-induced dynamic alternations also contribute to the heterogeneity of T cells in MM, which further make it difficult to obtain a clear view of T-cell status in relapsed and refractory MM patients. Indeed, ASCT is reported to induce dramatic changes, such as a persistent reduction in the CD4/CD8 T cell ratio and emergence of exhausted or senescent T cells.93-95 Again, in-depth profiling of MM-specific T cells is warranted to understand their functional status.
MM-associated immunosuppressive cells
Immunosuppressive myeloid cells, such as TAMs and myeloid-derived suppressor cells (MDSCs), are well-characterized subsets that potently suppress effector antitumor immunity.96 Growing evidence suggests that the proinflammatory MM niche orchestrated by damage-associated molecular patterns (DAMPs) and innate immune receptors has a strong impact on the generation and/or functional maturation of these myeloid cells. Hope et al showed that versican, one of the abundant ECM components in the MM niche, can be recognized by TLR-2 on monocytes/macrophages, leading to the generation of tolerogenic protumor macrophages.97 By contrast, its proteolytic product, versikine, can polarize macrophages into an immunogenic phenotype, which is partially independent of TLR-2,98 providing a novel cross talk between DAMPs and TAM polarization in the MM niche. More recently, Zavidij et al showed that BM CD14+ monocytes with reduced major histocompatibility complex class II expression have protumor functions and immunosuppressive activity, suggesting that precursors of TAMs are already educated to promote MM progression.83 In general, MDSCs are generated from BM myeloid progenitors in response to cytokines and growth factors, followed by mobilization into the TME in response to chemokines.96 Because MM cells grow predominantly in the BM, in situ MDSC generation can occur in response to MM-associated inflammation. Several lines of evidence support that S100A9 protein is a key DAMP that generates the proinflammatory niche and MDSCs by acting in an autocrine manner.99-101 The critical role of MDSCs is also supported by the transcriptional immune landscape of newly diagnosed MM patients, showing an inverse correlation between cytotoxic lymphocyte signature genes and MDSC-related genes.102 Although S100A9 induces proinflammatory signaling via cell surface pattern recognition receptors TLR-4 and RAGE, the cytosolic danger sensor called the inflammasome also drives MM progression by processing IL-1 family cytokines, especially IL-18.102 Inflammation-driven immunosuppressive myeloid cell subsets provide important therapeutic targets in the era of immunotherapy; however, further understanding of the heterogeneity and functional redundancy of myeloid subsets will be necessary to design the optimal therapeutic approach.
BM is a preferential site for migration and/or retention of Tregs, and the proportion of Tregs in CD4 T cells in BM are generally higher compared with in blood under steady-state conditions.84 Kawano et al recently characterized MM-associated Tregs and showed that activated Tregs expressing multiple immune checkpoint molecules were increased compared with control subjects. Mechanistically, tumor-derived type 1 IFN was implicated in shaping MM-associated Treg phenotypes.103 In general, depletion of tumor-associated Tregs has been recognized as a potential immunotherapeutic approach.104 In this context, depletion of CD38+ Tregs might be an additional mechanism of action by daratumumab,95,105 although the exact impact on antimyeloma immunity will require further investigation.
Soluble mediators
Several metabolites have also gained prominence as therapeutic targets in the immunosuppressive TME. Indoleamine 2,3-dioxygenase is an inducible enzyme that catalyzes tryptophan into kynurenine metabolites, and the indoleamine 2,3-dioxygenase–mediated deprivation of tryptophan and accumulation of kynurenine metabolites suppress antimyeloma immunity.106 In addition to MDSCs and TAMs, pDCs may also contribute to this metabolic immunosuppression, because the interaction between MM cells and pDCs was recently shown to upregulate kynurenine-3-mono-oxygenase, a downstream enzyme that can catabolize a tryptophan metabolite.107 Adenosine is a well-characterized immunosuppressive metabolite that is derived from extracellular adenosine triphosphate (catalyzed by CD39/CD73 ectoenzymes) and nicotinamide adenine dinucleotide (catalyzed by CD38/CD203a/CD73 ectoenzymes).108,109 Consistent with the fact that CD38 and CD39 are expressed on MM cells, elevated levels of adenosine were detected in active MM patients compared with healthy patients and those with MGUS.110 Of note, isatuximab and, to a much lesser extent, daratumumab have the ability to inhibit enzymatic activity of CD38.111 However, it remains unknown whether blockade of this CD38-dependent pathway can significantly reduce adenosine levels in the MM BM, because it is possible that CD39-induced adenosine might be sufficient to generate an adenosine-rich milieu. Several lines of evidence support that aberrant lipid metabolism supported by BM adipocytes fuels MM growth.112,113 However, it remains to be addressed whether altered lipid metabolism also induces immune reprograming in the MM niche. Inhibitory cytokines, such as TGF-β and IL-10, have been implicated in a broad range of malignancies, including MM.114 In preclinical solid malignancies, clustered regularly spaced palindromic repeats–mediated deletion of TGF-β receptor II in chimeric antigen receptor (CAR) T cells has shown good efficacy.115 This cell type–specific targeting approach (deleting in CAR T cells or blocking by multispecific antibody) may be feasible as MM immunotherapy, although further studies are necessary to determine the optimal therapeutic target.
Concluding remarks
Immunotherapy has emerged as a new pillar in MM treatment, and recently approved monoclonal antibodies against CD38 (daratumumab and isatuximab) and SLAMF7 (elotuzumab) have broadened therapeutic options as a result of their unique immunological mechanisms of action.116 More recently, CAR T-cell therapy against B-cell maturation antigen has shown remarkable clinical responses in relapsed or refractory MM patients.117 Also, bispecific T-cell engager antibodies against CD3 and B-cell maturation antigen are being actively tested in clinical trials.117,118 Although various immunotherapeutic approaches are being developed, it is still challenging to completely eliminate residual MM cells. Obviously, T-cell senescence acts as a major barrier for T-cell–based therapeutic approaches, and various strategies are being tested to reverse T-cell senescence.119 Given that malignant PCs require the BM niche for their survival, an in-depth understanding of the MM niche structure might also provide clues for better immune-mediated control. Although the importance of antitumor immunity in MM control is now appreciated, we have yet to obtain the complete picture of cancer immunoediting in PC dyscrasias. The complex interplay between tumor cells and the immune system can have bidirectional impacts: clonal evolution of tumor cells and alteration of the immune milieu. Additionally, a broad range of antimyeloma therapies (including, but not limited to, immunotherapy) can trigger a dynamic alteration of the immune environment. Given the heterogeneity of tumor cells and immune subsets, a personalized immune milieu profile might be necessary to identify the optimal targets to harness antimyeloma immunity. Indeed, the impact of tumor heterogeneity on the immune microenvironment remains largely unknown. Development of preclinical models that represent the heterogeneity of human MM will also be warranted as a research platform to investigate myeloma immunity and immunotherapy. Overall, a comprehensive understanding of the tumor immune microenvironment will provide clues to overcome therapeutic resistance.
Acknowledgments
K.N. is supported by the Naito Foundation and NHMRC Project Grant 1159593. M.J.S. is supported by a NH&MRC Investigator Grant (L3-T4, 1173958) and Program Grant (1132519). K.N. and M.J.S. were the recipients of a Leukemia Foundation of Australia SERP grant. L.M. is supported by a “Fondation ARC pour la Recherche sur le Cancer” Program Grant (ARC PGA1-20190208630), a Cancer Research Institute/Bristol Myers Squibb CLIP Grant, and the Institut National du Cancer (PLBIO R19-124).
Authorship
Contribution: K.N., M.J.S., and L.M. wrote and edited the manuscript.
Conflict-of-interest disclosure: M.J.S. has research agreements with Bristol Myers Squibb and Tizona Therapeutics and is a member of the Scientific Advisory Board for Tizona Therapeutics and Compass Therapeutics. L.M. has research agreements with Bristol Myers Squibb, Sanofi-Aventis, and Roche. K.N. declares no competing financial interests.
Correspondence: Kyohei Nakamura, QIMR Berghofer Medical Research Institute, 300 Herston Rd, Herston, QLD 4006, Australia; e-mail: kyohei.nakamura@qimrberghofer.edu.au; and Ludovic Martinet, INSERM UMR 1037, Cancer Research Center of Toulouse, 2 av Hubert Curien, 31037 Toulouse, France; e-mail: ludovic.martinet@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal