

In this issue of Blood, show that a tumor necrosis factor α (TNFα)-mediated pathway regulating interleukin-27 receptor subunit α (IL-27Ra) expression on hematopoietic stem cells (HSCs) mediates changes in HSCs that occur in old age, including myeloid skewing, reduced reconstitution potential, impaired self-renewal, and a proinflammatory phenotype.1
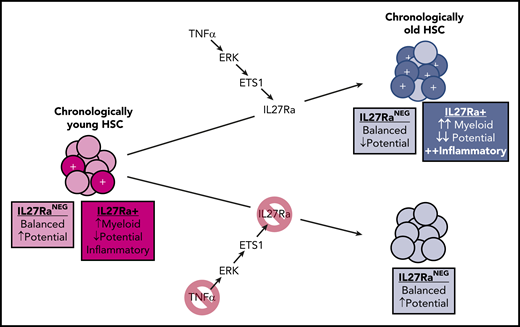
In young mice, a small fraction of HSCs are IL27Ra+, playing an important role in myeloid recovery following a microbial insult. In old age, a TNFα-ERK-ETS1 pathway upregulation leads to increased transcription of the IL-27Ra gene, the product of which promotes myeloid bias, an inflammatory phenotype, and reduced stem cell potential. Note that some reductions in stem cell potential occur with aging on top of the IL-27Ra–mediated effects.
In young mice, a small fraction of HSCs are IL27Ra+, playing an important role in myeloid recovery following a microbial insult. In old age, a TNFα-ERK-ETS1 pathway upregulation leads to increased transcription of the IL-27Ra gene, the product of which promotes myeloid bias, an inflammatory phenotype, and reduced stem cell potential. Note that some reductions in stem cell potential occur with aging on top of the IL-27Ra–mediated effects.
Sadly, our bodies and their constituent systems and tissues undergo decline in our later years, which is readily apparent in our hematopoietic system. Old humans, as well as other old animals such as mice, exhibit dramatic changes in blood cell production, with reduced HSC activity, reduced generation of lymphocytes, a bias toward myeloid cell production, and greater inflammatory cytokine production.2,3 These changes can negatively affect other tissues, as the chronic inflammation of old age is associated with multiple disease risks, including heart disease and cancers4,5 ; they can also greatly impair adaptive immune responses against pathogens. We have been tragically reminded of this last point during the ongoing COVID-19 pandemic, which is most deadly to the elderly.
He et al unraveled a novel TNFα-ERK-ETS1-IL27Ra pathway that contributes to aging phenotypes of HSCs. They show that TNFα, ERK, and ETS1 are each necessary (and TNF-α is sufficient) for the induction of IL27Ra in HSCs. Strikingly, old mice with IL27Ra knockout avoid most of these aging-associated changes in their HSCs (see figure). Although the number of HSCs activating this pathway greatly increases in old age, the authors found that even young mice exhibit some IL27Ra+ HSCs with these aging-like (albeit less pronounced) phenotypes, and that these cells are important for myeloid recovery following a microbial challenge. Similar distributions of IL27Ra+ HSCs are also evident in humans.
This TNFα-IL27Ra pathway is shown to likely play a protective role during microbial challenge, as modeled here using lipopolysaccharide challenge, promoting increased numbers of HSCs and a faster recovery of myeloid progenitors. Although increased TNFα expression seems to be most critical for activating this pathway in old age, IL-1β induced during an infection should also contribute to IL-27Ra regulation via NF-κB. Myeloid cells such as granulocytes are the frontline defense against pathogens, and replenishing these short-lived immune cells during and after an infection is critical for animal survival.3 Hence, it will be important to determine whether IL-27Ra is required for optimal animal survival during an actual infection. Moreover, results support the possibility that increased HSC numbers, increased fraction of IL27Ra+ HSCs, and myeloid skewing in old age are manifestations of normal pathogen-response programs operational throughout life but which become chronic in old age.
However, why do these programs, and the underlying inflammation, become chronic in old age? Although this aging-associated inflammation is often referred to as “sterile,” it may not be. Intestinal permeability has been shown to increase in old age,6 and aging-associated increases in TNFα levels are necessary for this increased permeability.7 A positive feedback loop between gut dysbiosis in old age, systemic entry of bacterial products, and increased inflammatory cytokines such as TNFα can at least in part explain chronic inflammatory changes in old age.7 He et al suggest another positive feedback loop, with increases in TNFα→IL-27Ra leading to greater immunosenescence and myeloid skewing, which itself leads to increased TNFα production from myeloid cells. But why would the chronic manifestation of these feedback mechanisms be mostly restricted to old age? Connecting the previous intestinal studies with the current research by He et al, one can surmise that TNFα/IL-27Ra–dependent changes in HSCs in old age indeed reflect a chronic manifestation, via intestinal permeability–facilitated entry of bacterial products, of the same antimicrobial responses that limit disease incidence throughout life. Humans and other animals simply did not evolve to live forever, and investments in tissue maintenance wane at ages at which the odds of contributing to future generations are historically low. Such investments include maintenance of our intestinal tract and its critical barrier function.
What on the surface seems to be contradictory, an important defense pathway that contributes to aging phenotypes later in life, actually makes a lot of sense. The concept of antagonistic pleiotropy was proposed >60 years ago by Williams8 : genetically encoded phenotypes (or programs) that contribute to animal fitness in youth but which also promote aging phenotypes and reduced survival in old age are still favored by natural selection as long as, in balance, animal fitness (reproductive success) is increased. Because most animals in the wild do not survive to ages at which senescent phenotypes are evident, natural selection acts to maximize survival and reproductive success in youth even when these same programs contribute to our eventual demise in old age. Although the old mice studied by He et al were aged >2 years, mice rarely survive past 1 year in the wild, given high extrinsic hazards such as resource limitations, predation, disease, and cold. Only the luckiest, and rarest, wild mouse will die of “old age.”
The million-dollar question of course is: is there anything we can do about these aging-related changes? Although the simple solution might seem to be to block inflammation in old age, we need to keep in mind this antagonistic pleiotropy, that inflammation is important for our defenses. In fact, while blocking IL-1β in people resulted in decreases in cancers and heart disease, overall survival was not improved due to increases in deaths by infections.9 Still, a more mechanistic and detailed understanding of the pathways controlling functional decline in old age, as provided for HSCs by He et al, could lead to more nuanced and targeted strategies to mitigate these declines without overly compromising the evolved functions of the pathways underlying these aging-associated perturbations.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal