

Although mitochondria are known to be a major source of reactive oxygen species (ROS), and oxidative stress is thought to contribute to pathology in sickle cell disease, in this issue of Blood, have shown that arginine therapy can improve mitochondrial function and decrease oxidative stress.1
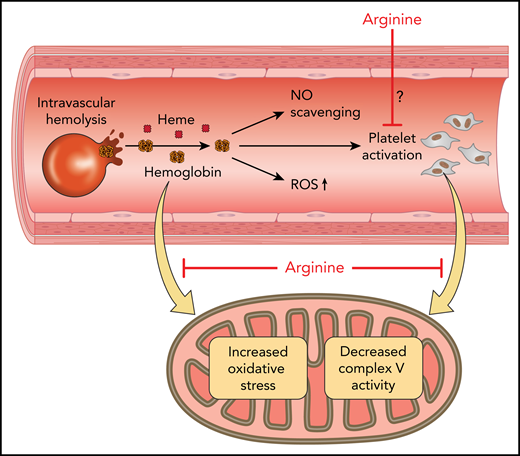
Mitochondrial therapeutics. Red cell hemolysis produces cell-free hemoglobin and heme, which leads to NO scavenging, formation of ROS, and platelet activation. Red cells also release ADP, which activates platelets and arginase, decreasing NO bioavailability (not shown). Hemoglobin leads to decreased platelet mitochondrial complex V activity and oxidative stress. Arginine therapy improves mitochondrial function in sickle cell patients, but whether the mechanism involves direct inhibition of platelet activation by NO, countering the deleterious effects of hemoglobin/heme, transiently modulating platelet complex I, or by limiting systemic-ischemia-reperfusion stress remains to be determined. Professional illustration by Patrick Lane, ScEYEnce Studios.
Mitochondrial therapeutics. Red cell hemolysis produces cell-free hemoglobin and heme, which leads to NO scavenging, formation of ROS, and platelet activation. Red cells also release ADP, which activates platelets and arginase, decreasing NO bioavailability (not shown). Hemoglobin leads to decreased platelet mitochondrial complex V activity and oxidative stress. Arginine therapy improves mitochondrial function in sickle cell patients, but whether the mechanism involves direct inhibition of platelet activation by NO, countering the deleterious effects of hemoglobin/heme, transiently modulating platelet complex I, or by limiting systemic-ischemia-reperfusion stress remains to be determined. Professional illustration by Patrick Lane, ScEYEnce Studios.
Sickle cell disease is caused by a mutant form of hemoglobin that polymerizes upon deoxygenation, producing rigid red blood cells that are prone to hemolysis. Hemolytic anemia releases cell-free hemoglobin that scavenges the important signaling molecule nitric oxide (NO), directly generates ROS, activates oxidases that produce ROS, and denatures to release heme, which activates innate inflammatory signaling pathways.2 Hemolysis also releases arginase-1, which converts the substrate for NO synthase, arginine, to ornithine and thereby contributes to the NO deficiency seen in sickle cell disease.3 Therefore, arginine administration has been explored as a therapeutic based on its potential to restore NO bioavailability, and clinical efficacy has been demonstrated in small randomized clinical trials.4 The new study by Morris et al suggests that, like another recently approved therapeutic amino acid, glutamine,5 arginine may reduce oxidative stress in sickle cell disease (see figure).
Mitochondria are known to be a major source of ROS; in sickle cell disease, many investigators have shown that ROS is generated from the mitochondria and other oxidases, including xanthine oxidase, NADPH oxidase, oxidized hemoglobin, and uncoupled endothelial NO synthase.6 Prior studies have shown that in patients with sickle cell disease, hemolysis-derived hemoglobin activates platelets, increases ROS generation from platelet mitochondria, and oxidatively impairs complex V (ATPase) function.7 These data suggest that platelet activation by hemolysis in sickle cell disease is related to a mitochondrial oxidative enzymopathy, with activation associated with complex V oxidative impairment. The isolation and study of platelet mitochondria allow for a mitochondrial biopsy, with recent studies suggesting that platelet mitochondria may serve as a biomarker reflecting systemic mitochondrial function.8 In the current study, Morris et al have extended these data and now show that systemic IV arginine therapy in patients with sickle cell disease vasoocclusive episodes (VOEs) can improve mitochondrial complex V function and decrease ROS formation. Upon hospital admission, mitochondrial complex V activity was found to be lower than that previously found for patients with sickle cell disease at steady state.7 Arginine infusion in 12 pediatric patients with VOEs improved mitochondrial complex IV and V activity and decreased markers of oxidative stress (protein carbonyls and malondialdehyde). The improvements in mitochondrial function were greatest when the arginine infusions included a loading dose.
From a clinical perspective, a potentially important finding of the current study is that the IV administered arginine was most effective at improving mitochondrial function when a loading dose was included. There were 3 study groups, 2 receiving a loading arginine dose followed by either continuous infusion or infusion 3 times a day and the third receiving infusion 3 times a day with no loading dose. Comparisons were made between samples obtained upon hospital admission and upon discharge. Although only 4 patients were evaluated with each dosing scheme, the improvements in mitochondrial complex IV and V activity were more robust with inclusion of a loading dose; improvements in oxidative stress were similar in all groups. Furthermore, the infusion doses used in this study can potentially inform larger clinical trials. Future studies will be needed to test whether oxidative stress and mitochondrial function improve on their own without arginine infusion during VOE resolution, because no placebo group was evaluated.
There are multiple mechanistic questions raised by the study by Morris et al. Several credible mechanisms have been published evaluating how hemolysis and hemoglobin activate platelets, including ADP release from red blood cells, ROS generation from hemoglobin, and NO scavenging.9 Inhibition of complex V has been shown to activate platelets,7 but it is still unknown whether platelets activated by various agonists (eg, ADP, collagen, and thrombin) have compromised mitochondrial function. Therefore, the mechanism of action of hemoglobin on mitochondrial activity remains to be elucidated. In addition, the mechanism for how arginine improves mitochondrial function in vivo during VOEs, demonstrated in the current study, is not known. Arginine is metabolized to NO, which is known to inhibit platelet activation, reversibly inhibit mitochondrial respiration by binding to cytochrome C oxidase, and potentiate mitochondrial biogenesis. Prior studies have also demonstrated that NO and nitrite, formed from oxidation of NO, can reversibly S-nitrosate complex I of the mitochondrial electron transport chain during ischemia and reperfusion injury.10 This transient inhibition of complex I can then prevent downstream oxidation of aconitase and inhibition of complex IV. However, in the current study, there was no observed inhibition of mitochondrial respiration or increased mitochondrial biogenesis. Another possibility is that improved regional blood flow and limited regional ischemia reperfusion inflammation and ROS generation modulated platelets indirectly, by limiting activation in injured vascular beds. More studies with pharmacodynamic measurements of mitochondrial function will be needed to understand how arginine and NO modulate mitochondrial complex activity.
In conclusion, this translational study advances potentially important concepts: (1) mitochondrial dysfunction contributes to human disease, including sickle cell disease, and (2) improving mitochondrial function with arginine therapy may represent an effective treatment strategy to reverse oxidative mitochondrial enzymopathy. The study also provides insights into dosing schemes for future clinical trials and encourages several in vitro mechanistic studies on the interplay of NO, hemoglobin, platelets, and mitochondria.
Conflict-of-interest disclosure: M.T.G. and D.B.K.-S. are coinventors on patents related to hemolysis and directed to the use of nitrite salts in cardiovascular diseases. M.T.G. is a coinvestigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguat as a treatment for patients with sickle cell disease.
