

Key Points
Whole-exome sequencing of a large series of BI-ALCL demonstrates recurrent mutations in epigenetic regulators.
An accumulation of alterations in epigenetic modifiers and genes in the JAK/STAT pathway likely drives BI-ALCL oncogenesis.

Abstract
The oncogenic events involved in breast implant–associated anaplastic large cell lymphoma (BI-ALCL) remain elusive. To clarify this point, we have characterized the genomic landscape of 34 BI-ALCLs (15 tumor and 19 in situ subtypes) collected from 54 BI-ALCL patients diagnosed through the French Lymphopath network. Whole-exome sequencing (n = 22, with paired tumor/germline DNA) and/or targeted deep sequencing (n = 24) showed recurrent mutations of epigenetic modifiers in 74% of cases, involving notably KMT2C (26%), KMT2D (9%), CHD2 (15%), and CREBBP (15%). KMT2D and KMT2C mutations correlated with a loss of H3K4 mono- and trimethylation by immunohistochemistry. Twenty cases (59%) showed mutations in ≥1 member of the JAK/STAT pathway, including STAT3 (38%), JAK1 (18%), and STAT5B (3%), and in negative regulators, including SOCS3 (6%), SOCS1 (3%), and PTPN1 (3%). These mutations were more frequent in tumor-type samples than in situ samples (P = .038). All BI-ALCLs expressed pSTAT3, regardless of the mutational status of genes in the JAK/STAT pathway. Mutations in the EOMES gene (12%) involved in lymphocyte development, PI3K-AKT/mTOR (6%), and loss-of-function mutations in TP53 (12%) were also identified. Copy-number aberration (CNA) analysis identified recurrent alterations, including gains on chromosomes 2, 9p, 12p, and 21 and losses on 4q, 8p, 15, 16, and 20. Regions of CNA encompassed genes involved in the JAK/STAT pathway and epigenetic regulators. Our results show that the BI-ALCL genomic landscape is characterized by not only JAK/STAT activating mutations but also loss-of-function alterations of epigenetic modifiers.
Introduction
Breast implant–associated anaplastic large-cell lymphoma (BI-ALCL) is a rare form of T-cell lymphoma arising adjacent to a breast implant that was recently recognized as a provisional entity in the 2017 revised World Health Organization lymphoma classification.1 The incidence of BI-ALCL varies from 1 in 30 000 to 1 in 1000 women with breast implants,2-5 with ∼660 cases reported to the US Food and Drug Administration.4
Most BI-ALCL patients (80%) present with an isolated effusion adjacent to the implant (seroma BI-ALCL) and have an excellent outcome.5-7 In contrast, a minority of patients (20%) present with a breast tumor that might disseminate outside the breast and have a worse prognosis.5-7 We recently described 2 BI-ALCL histological subtypes that correlate with the clinical presentations: (1) in situ BI-ALCL, which is found in patients with seroma and characterized by lymphoma cells lining the capsule border and/or suspended in a serous/fibrinoid material; and (2) tumor-type BI-ALCL, which is mostly found in patients with tumor mass and characterized by capsule invasion.6,8
Although BI-ALCLs share a similar morphology and immunophenotype with systemic ALCLs, they lack rearrangements involving ALK, DUSP22, and TP63 genes.8,9 In a manner reminiscent of systemic ALCL, a few studies using next-generation sequencing (NGS) have pointed toward a putative dysregulation of JAK/STAT signaling in BI-ALCL oncogenesis. Recurrent somatic STAT3 mutations have been indeed observed in 26% to 64% of analyzed cases.6,9-15 However, since only 2 BI-ALCL cases have been thoroughly studied by whole-exome sequencing (WES), the mutational landscape of this entity has not yet been reported.
To further explore potential molecular mechanisms involved in BI-ALCL pathogenesis, we have analyzed a large set of 34 BI-ALCL cases by WES and/or targeted deep sequencing (TDS). The mutational landscape of BI-ALCL showed both JAK/STAT signaling dysregulation and epigenetic alterations.
Materials and methods
Cases selection and tumor samples
Fifty-four BI-ALCL cases (including 2 BI-ALCL cases from Belgium pathology departments at Centre Hospitalier Universitaire de Liège and Centre Hospitalier Universitaire UCLouvain Namur) were collected between 2010 and May 2018 via the French Lymphopath network, aiming at providing a systematic review of every newly diagnosed or suspected lymphoma.16 Among them, 34 cases were selected based on matched germline samples availability and/or tumor DNA quality for downstream genomic analysis. All of these cases were further reviewed by 4 of the coauthors (Camille Laurent, A.N., P.G., and L.X.). They were histologically subclassified as in situ or tumor-type BI-ALCL and staged according to the MD Anderson TNM classification7,17,18 based on the pathological material submitted to the Lymphopath network. Clinical and pathological features are detailed in Table 1. Based on hematoxylin and eosin sections and appropriate immunostaining, a semiquantitative estimation of tumor cell content was given as ≤20%, 20% to 50%, and >50% tumor cells. In addition, 20 cases with available remaining DNA were analyzed for their T-cell receptor (TCR) repertoire profile using a CapTCR-seq NGS technique (see details in supplemental Methods, available on the Blood Web site) in order to evaluate the proportion of neoplastic T cells in comparison with the total T-cell population (number of reads corresponding to the dominant clonal TCRβ gene rearrangement among the total number of reads with TCRβ gene rearrangements) (supplemental Table 1).
Main clinical characteristics and histological features of 34 BI-ALCL patients
| No. | Age, y | BI-ALCL histological subtype (T component of MD Anderson TNM staging) | Implant characteristics | Ann Arbor staging | Therapy (implant removal/CT/RT) | Overall survival (mo) (outcome NED/DOD/DOC) | T-cell clone (% tumor cells per sample) | Exome sequencing by WES/TDS | ||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of implant surgeries prior to BI-ALCL diagnosis (reason for implant) | ≥1 macrotextured implant | Delay from the last implant to Bi-ALCL diagnosis (delay from first implant), y | ||||||||
| 2 | 42 | In situ (T1) | 2 (cosmetic) | Yes | 10.5 (20) | I | Implant removal + RT | 56.48 (NED) | Yes (≤20) | WES |
| 5 | 54 | In situ (T1) | NA (cosmetic) | NA | 9 (NA) | IE | NA | NA | NA (20-50) | WES |
| 6 | 62 | In situ (T1) | 2 (breast carcinoma) | Yes | 0.26 (17) | IV | Implant removal | 55.59 (NED) | Yes (>50) | WES |
| 7 | 76 | In situ (T1) | 1 (breast carcinoma) | Yes | 14.23 | I | Implant removal | 44.91 (NED) | Yes (≤20) | WES + TDS |
| 8 | 43 | In situ (T1) | 1 (breast carcinoma) | Yes | 6.7 | I | Implant removal | 42.81 (NED) | Yes (20-50) | WES + TDS |
| 10 | 61 | Tumor type (T4) | 4 (cosmetic) | Yes | 7.2 (17) | IV | Implant removal + CT1 | 77.24 (NED) | Yes (>50) | TDS |
| 11 | 54 | Tumor type (T4) | NA (cosmetic) | Yes | 3 (NA) | II | NA | NA | Yes (>50) | TDS |
| 13 | 82 | Tumor type (T4) | 1 (breast carcinoma) | Yes | 6.96 | IV | Implant removal + CT1 | 10.45 (DOD) | Yes (>50) | WES |
| 14 | 80 | Tumor type (T3) | NA (cosmetic) | Yes | 8 | IE | Implant removal + CT1 | 3 (NED) | NA (>50) | TDS |
| 15 | 50 | In situ (T1) | NA (cosmetic) | Yes | 8 | IE | Implant removal | NA | Yes (≤20) | TDS |
| 16 | 77 | In situ (T1) | 3 (breast carcinoma) | Yes | 1.23 (NA) | I | Implant removal + CT2 | 60.29 (NED) | NA (≤20) | WES |
| 17 | 74 | Tumor type (T4) | 2 (breast carcinoma) | Yes | 8.12 (NA) | IV | Implant removal + CT1 | 44.88 (NED) | Yes (≤20) | WES |
| 18 | 75 | In situ (T1) | 5 (breast carcinoma) | Yes | 1.27 (NA) | IV | Implant removal + CT1 | 16.39 (DOD) | NA (≤20) | TDS |
| 19 | 57 | Tumor type (T4) | 2 (breast carcinoma) | Yes | 7.57 (NA) | IV | NA | 3.71 (DOC) | Yes (20-50) | WES + TDS |
| 21 | 62 | In situ (T1) | 1 (breast carcinoma) | Yes | 9.37 | I | Implant removal | 27.73 (NED) | Yes (≤20) | WES + TDS |
| 22 | 39 | In situ (T1) | 1 (cosmetic) | Yes | 6.48 | I | Implant removal | 23.95 (NED) | NA (20-50) | WES |
| 24 | 62 | In situ (T1) | 2 (breast carcinoma) | NA | 4.57 (NA) | I | Implant removal + CT | 30.39 (NED) | NA (≤20) | WES + TDS |
| 26 | 54 | In situ (T1) | 1 (cosmetic) | Yes | 9.88 | I | Implant removal | 27.17 (NED) | NA (≤20) | TDS |
| 28 | 66 | Tumor type (T2) | 2 (breast carcinoma) | Yes | 5.36 (NA) | II | Implant removal + CT | 22.01 (NED) | No (≤20) | WES + TDS |
| 29 | 44 | Tumor type (T2) | 1 (cosmetic) | Yes | 8.07 | I | Implant removal | 22.64 (NED) | No (20-50) | WES + TDS |
| 30 | 54 | Tumor type (T2) | 2 (breast carcinoma) | Yes | 3.6 (NA) | I | Implant removal + CT | 25.26 (NED) | No (>50) | WES + TDS |
| 31 | 54 | In situ (T1) | 1 (cosmetic) | Yes | 5.11 | IV | Implant removal | 24.67 (NED) | Yes (≤20) | TDS |
| 32 | 65 | In situ (T1) | 3 (cosmetic) | Yes | 4.25 (NA) | I | Implant removal | 21.32 (NED) | Yes (≤20) | WES |
| 33 | 40 | Tumor type (T3) | 1 (cosmetic) | Yes | 10.12 | IV | Implant removal + CT | 19.22 (NED) | Yes (>50) | WES + TDS |
| 34 | 50 | Tumor type (T3) | 1 (cosmetic) | Yes | 9.85 | I | Implant removal | 15.74 (NED) | Yes (>50) | WES + TDS |
| 35 | 43 | Tumor type (Tx) | 1 (cosmetic) | NA | 12.31 | IV | Implant removal + CT | 23.49 (NED) | Yes (>50) | TDS |
| 36 | 72 | In situ (T1) | 1 (cosmetic) | Yes | 22.77y | I | Implant removal | 11.76 (NED) | Yes (>50) | TDS |
| 37 | 63 | Tumor type (T3) | 1 (breast carcinoma) | Yes | 8.74y | I | CT | 7.75 (DOD) | NA (>50) | WES |
| 38 | 55 | In situ (T1) | 3 (breast carcinoma) | Yes | 5.31 (NA) | I | Implant removal | 13.11 (NED) | Yes (>50) | TDS |
| 42 | 48 | In situ (T1) | 2 (cosmetic) | Yes | 4.88 (NA) | I | Implant removal | 13.54 (NED) | Yes (>50) | WES + TDS |
| 48 | 65 | Tumor type (T4) | 3 (cosmetic) | Yes | 0.5 (NA) | I | Implant removal | 7.66 (NED) | Yes (≤20) | TDS |
| 50 | 58 | In situ (T1) | 2 (cosmetic) | Yes | 1.89 (11) | IV | Implant removal | NA | No (≤20) | TDS |
| Bel1 | 59 | In situ (T1) | 2 (NA) | Yes | 8 (NA) | I | Implant removal | 18 (NED) | NA (>50) | WES |
| Bel2 | 47 | Tumor type (T2) | 2 (NA) | Yes | 6 (13) | NA | Implant removal | 22 (NED) | Yes (20-50) | WES + TDS |
| No. | Age, y | BI-ALCL histological subtype (T component of MD Anderson TNM staging) | Implant characteristics | Ann Arbor staging | Therapy (implant removal/CT/RT) | Overall survival (mo) (outcome NED/DOD/DOC) | T-cell clone (% tumor cells per sample) | Exome sequencing by WES/TDS | ||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of implant surgeries prior to BI-ALCL diagnosis (reason for implant) | ≥1 macrotextured implant | Delay from the last implant to Bi-ALCL diagnosis (delay from first implant), y | ||||||||
| 2 | 42 | In situ (T1) | 2 (cosmetic) | Yes | 10.5 (20) | I | Implant removal + RT | 56.48 (NED) | Yes (≤20) | WES |
| 5 | 54 | In situ (T1) | NA (cosmetic) | NA | 9 (NA) | IE | NA | NA | NA (20-50) | WES |
| 6 | 62 | In situ (T1) | 2 (breast carcinoma) | Yes | 0.26 (17) | IV | Implant removal | 55.59 (NED) | Yes (>50) | WES |
| 7 | 76 | In situ (T1) | 1 (breast carcinoma) | Yes | 14.23 | I | Implant removal | 44.91 (NED) | Yes (≤20) | WES + TDS |
| 8 | 43 | In situ (T1) | 1 (breast carcinoma) | Yes | 6.7 | I | Implant removal | 42.81 (NED) | Yes (20-50) | WES + TDS |
| 10 | 61 | Tumor type (T4) | 4 (cosmetic) | Yes | 7.2 (17) | IV | Implant removal + CT1 | 77.24 (NED) | Yes (>50) | TDS |
| 11 | 54 | Tumor type (T4) | NA (cosmetic) | Yes | 3 (NA) | II | NA | NA | Yes (>50) | TDS |
| 13 | 82 | Tumor type (T4) | 1 (breast carcinoma) | Yes | 6.96 | IV | Implant removal + CT1 | 10.45 (DOD) | Yes (>50) | WES |
| 14 | 80 | Tumor type (T3) | NA (cosmetic) | Yes | 8 | IE | Implant removal + CT1 | 3 (NED) | NA (>50) | TDS |
| 15 | 50 | In situ (T1) | NA (cosmetic) | Yes | 8 | IE | Implant removal | NA | Yes (≤20) | TDS |
| 16 | 77 | In situ (T1) | 3 (breast carcinoma) | Yes | 1.23 (NA) | I | Implant removal + CT2 | 60.29 (NED) | NA (≤20) | WES |
| 17 | 74 | Tumor type (T4) | 2 (breast carcinoma) | Yes | 8.12 (NA) | IV | Implant removal + CT1 | 44.88 (NED) | Yes (≤20) | WES |
| 18 | 75 | In situ (T1) | 5 (breast carcinoma) | Yes | 1.27 (NA) | IV | Implant removal + CT1 | 16.39 (DOD) | NA (≤20) | TDS |
| 19 | 57 | Tumor type (T4) | 2 (breast carcinoma) | Yes | 7.57 (NA) | IV | NA | 3.71 (DOC) | Yes (20-50) | WES + TDS |
| 21 | 62 | In situ (T1) | 1 (breast carcinoma) | Yes | 9.37 | I | Implant removal | 27.73 (NED) | Yes (≤20) | WES + TDS |
| 22 | 39 | In situ (T1) | 1 (cosmetic) | Yes | 6.48 | I | Implant removal | 23.95 (NED) | NA (20-50) | WES |
| 24 | 62 | In situ (T1) | 2 (breast carcinoma) | NA | 4.57 (NA) | I | Implant removal + CT | 30.39 (NED) | NA (≤20) | WES + TDS |
| 26 | 54 | In situ (T1) | 1 (cosmetic) | Yes | 9.88 | I | Implant removal | 27.17 (NED) | NA (≤20) | TDS |
| 28 | 66 | Tumor type (T2) | 2 (breast carcinoma) | Yes | 5.36 (NA) | II | Implant removal + CT | 22.01 (NED) | No (≤20) | WES + TDS |
| 29 | 44 | Tumor type (T2) | 1 (cosmetic) | Yes | 8.07 | I | Implant removal | 22.64 (NED) | No (20-50) | WES + TDS |
| 30 | 54 | Tumor type (T2) | 2 (breast carcinoma) | Yes | 3.6 (NA) | I | Implant removal + CT | 25.26 (NED) | No (>50) | WES + TDS |
| 31 | 54 | In situ (T1) | 1 (cosmetic) | Yes | 5.11 | IV | Implant removal | 24.67 (NED) | Yes (≤20) | TDS |
| 32 | 65 | In situ (T1) | 3 (cosmetic) | Yes | 4.25 (NA) | I | Implant removal | 21.32 (NED) | Yes (≤20) | WES |
| 33 | 40 | Tumor type (T3) | 1 (cosmetic) | Yes | 10.12 | IV | Implant removal + CT | 19.22 (NED) | Yes (>50) | WES + TDS |
| 34 | 50 | Tumor type (T3) | 1 (cosmetic) | Yes | 9.85 | I | Implant removal | 15.74 (NED) | Yes (>50) | WES + TDS |
| 35 | 43 | Tumor type (Tx) | 1 (cosmetic) | NA | 12.31 | IV | Implant removal + CT | 23.49 (NED) | Yes (>50) | TDS |
| 36 | 72 | In situ (T1) | 1 (cosmetic) | Yes | 22.77y | I | Implant removal | 11.76 (NED) | Yes (>50) | TDS |
| 37 | 63 | Tumor type (T3) | 1 (breast carcinoma) | Yes | 8.74y | I | CT | 7.75 (DOD) | NA (>50) | WES |
| 38 | 55 | In situ (T1) | 3 (breast carcinoma) | Yes | 5.31 (NA) | I | Implant removal | 13.11 (NED) | Yes (>50) | TDS |
| 42 | 48 | In situ (T1) | 2 (cosmetic) | Yes | 4.88 (NA) | I | Implant removal | 13.54 (NED) | Yes (>50) | WES + TDS |
| 48 | 65 | Tumor type (T4) | 3 (cosmetic) | Yes | 0.5 (NA) | I | Implant removal | 7.66 (NED) | Yes (≤20) | TDS |
| 50 | 58 | In situ (T1) | 2 (cosmetic) | Yes | 1.89 (11) | IV | Implant removal | NA | No (≤20) | TDS |
| Bel1 | 59 | In situ (T1) | 2 (NA) | Yes | 8 (NA) | I | Implant removal | 18 (NED) | NA (>50) | WES |
| Bel2 | 47 | Tumor type (T2) | 2 (NA) | Yes | 6 (13) | NA | Implant removal | 22 (NED) | Yes (20-50) | WES + TDS |
CT, chemotherapy not specified; CT1, cyclophosphamide, Adriamycin, vincristine, and prednisone (6 cures) (CHOP or CHOP-like); CT2, dexamethasone, high-dose aracytine, oxaliplatin (DHAOX); DOC, dead from other cause (breast carcinoma); DOD, dead of disease; NA, not available; NED, no evidence of disease; RT, radiotherapy.
These patients were included in the framework of a T-cell lymphoma consortium (Tenomic), which was approved by the local ethical committee (Comité de Protection des Personnes Ile de France IX 08-009). Informed consent was obtained from patients for the analysis of tumor and germline specimens. Tissue samples were collected and processed following standard ethical procedures (Declaration of Helsinki 1975). Clinical and pathological features of 17 cases have been previously reported.8
Immunohistochemistry (IHC)
Immunohistochemical studies were performed on 3-µm-thick formalin-fixed paraffin-embedded (FFPE) sections using routine protocols on fully automated platforms (Ventana, Benchmark XT, Tucson, AZ). For diagnostic purposes, the cases were tested for a wide panel of antibodies detailed in supplemental Table 2. To functionally validate the mutations affecting STAT3 and STAT5B genes, antibodies against phospho-STAT3 (clone D3A7, 1:50; Cell Signaling Technology, Beverly, MA) and phospho-STAT5 (clone E208, 1:100; Abcam, Cambridge, United Kingdom) were used. The histone H3K4 methylation status was evaluated using antibodies against histone‐3 lysine‐4 (H3K4)me3 (trimethyl Lys4, polyclonal GTX128954, 1:100; GeneTex, Irvine, CA) and H3K4me1 (ab8895 clone, 1:500; Abcam). Computer-assisted software was used to score histone H3K4 methylation status (see details in supplemental Methods).
DNA extraction
Genomic tumor DNA was extracted from 10-µm-thick FFPE tissue sections using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA) or Maxwell 16 FFPE Plus LEV DNA Purification Kit (Promega) according to the manufacturers’ recommendations. To enrich in tumor cell content, a macrodissection was performed for 8 cases. For the cases analyzed by WES, paired germline DNA was extracted from either peripheral blood (n = 14) or macrodissected nontumoral FFPE capsular tissue (n = 8).
WES
WES was performed on 22 samples, including 12 in situ and 10 tumor-type BI-ALCL tissues and their matched germline DNA. DNA libraries were prepared using NEBU ultra V2 and Agilent SureSelect Clinical Research Exome V2. Sequencing was performed on an Illumina HiSeq4000 with an expected mean depth of 200× for the tumor and 70× for the germline. Sequences analysis was performed using Bcbio-Nextgen somatic variant calling pipeline (version 1.0.7a). Sequences were aligned to the human genome (version GRCh37) with Burrows-Wheeler Aligner software (version 0.7.17). Single-nucleotide variants (SNVs) and indels were detected with the combination of 4 algorithms (Mutect2 version 3.8,19 Vardict version 1.5,20 Strelka2,21 and Freebayes version 1.1.04622 ). Variants were annotated using VEP (version 94) and filtered as follows: coverage in tumor and corresponding normal samples ≥15, alternative bases coverage ≥3, and variant allele frequency (VAF) >2% (ratio between alternative allele reads and total reads). Variants described in the 1000 Genomes Project, gnomAD database, and ExAC database in >1% of the population were discarded. The calls with reads showing strong bias or present in the majority of the samples were filtered out. All reported variants passed visual inspection using the Integrative Genomics Viewer.
CNA analysis
Analysis of copy-number aberrations (CNAs) for paired tumor-normal whole-exome–sequenced samples was performed using the FACETS algorithm.23 The critical value for segmentation was set to 150. Other parameters remain the same as default values. To estimate the status calling, we extracted the estimated total copy number (under column tcn.em) and the ploidy from the segmentation files produced by FACETS. We also estimated the genotype to identify regions of copy-neutral loss of heterozygosity. Amplification was defined as >4 DNA copies.
TDS
To validate WES results, 24 BI-ALCL (12 in situ and 12 tumor-type cases) were analyzed using a TDS approach, with 500× average depth (497× [265-698×]); 12 of them were initially analyzed by WES, and 12 additional cases were used. The 406 genes FundationOne Heme panel used includes genes previously reported to be mutated in T-cell lymphomas and members of cytokine signaling and TCR signaling pathways (https://assets.ctfassets.net/vhribv12lmne/zBxaQC12cScqgsEk8seMO/a70860fea48927c7ff8b70e90c292182/F1H_TechnicalInformation.pdf).
Reads were aligned on the human genome (GRCh37) with BWA (version 0.7.17). Variant calling was done with Vardict (version 1.5) and GATK Haplotype Caller (GATK 3.8) algorithms. They were filtered on their allele frequency (VAF >2%), strand bias, depth (>50), and depth of the alternative allele (>3). Variants were annotated with VEP (version 94). The variants presented in a pool of normal based on WES analysis were discarded. Variants known as polymorphic were eliminated (variants known in 1000 Genomes, ExAC, or gnomAD with a frequency >1%). Only the variants associated with a frameshift, in-frame indel, start/stop codon change, or missense change were kept. The sequence alignments for all JAK/STAT variants were visually inspected and artifacts excluded.
Results
Clinical data and pathological features
Patients’ age at diagnosis ranged from 39 to 82 years. Thirty-two breast implants were textured, and the remaining 2 cases were of unknown status. Seventeen patients had experienced ≥2 implant surgeries before BI-ALCL diagnosis, with a median interval time between the last implant surgery and BI-ALCL diagnosis of 7.3 years (range, 6 to 273 months). Twenty-three patients (70%) were Ann Arbor stage I/II, and 30% were stage IV. Regarding the histological subtype, 19 patients had an in situ BI-ALCL (T1 stage according to MD Anderson TNM staging),7,17,18 and 15 had a tumor-type BI-ALCL (4 corresponding to T2 stage, 4 to T3 stage, 6 to T4 stage, and 1 nonevaluable). The main clinical and pathological characteristics of the 34 BI-ALCL patients are given in Table 1. By IHC, all cases were strongly and uniformly positive for CD30 and showed incomplete T-cell phenotype, with a common activated cytotoxic profile. Neoplastic cells were often positive for epithelial membrane antigen (90%), and ALK1 was consistently negative. Most patients had a favorable outcome, except 3 patients who died of lymphoma progression (cases 13, 18, and 37).24
Genomic landscape of BI-ALCL
WES was performed on 22 paired tumor/normal samples. The mean sequencing depth was 131× (84-153×) for the tumor and 75× (47×-145×) for the germline DNA. All but 3 BI-ALCLs showed somatic mutations in genes considered to be relevant to oncogenesis according to our bioinformatics filtering. In total, we identified 1248 nonsynonymous somatic mutations involving 1118 genes after variant filtering. SNVs represented 86% of the mutations (1069 SNVs for 979 genes). Among them, missense mutations were the most frequent alterations (88%), followed by nonsense (7%) and splice-site mutations (4%) (supplemental Figure 1A). Indels represented 14% of somatic mutations (179 mutations involving 153 genes), including 74% frameshift and 26% in-frame insertions or deletions. Among single-nucleotide mutations, G>T and C>A transversions were the most common (56%), followed by G>A and C>T transitions (30%), consistent with similar observations in other cancers and lymphomas.25 The mean count of nonsynonymous somatic alterations was 57, with a minimum of 9 and a maximum of 193 (see supplemental Table 3 for the complete list of mutations). The mutational landscape of BI-ALCL was remarkably homogeneous with enriched clusters among JAK-STAT, epigenetic modifier genes, and cell cycle or apoptosis pathways (supplemental Figure 1B).
JAK/STAT pathway activation in BI-ALCL
The most common alterations observed in BI-ALCL samples involved members of the JAK/STAT pathway, found in 64% (n = 14/22) of WES analyzed cases. STAT3 and JAK1 were the most frequently mutated genes, accounting for 41% (n = 9/22) and 18% (n = 4/22) of cases, respectively; these were followed by SOCS3 (9%), SOCS1 (5%), PTPN1 (5%), and STAT5B (5%). Interestingly, these mutations were not mutually exclusive, and co-occurrence of mutations in 2 genes of the JAK/STAT pathway was found in 4 cases (supplemental Figure 1B). All STAT3 mutations were missense mutations and occurred at p.S614R, p.G618R, p.D661Y, and p.I659L hotspots. Except for 1 nonsense mutation (p.R174*), the JAK1 mutations were missense and related to the p.G1097D/V/C/S hotspots. All of these aberrations have been previously shown to be gain-of-function mutations resulting in JAK/STAT pathway activation in various types of T-cell lymphoma.26,27
Highly recurrent alterations in epigenetic modifiers in BI-ALCL
We detected frequent mutations of epigenetic regulators and histone modifiers. Genes encoding H3K4 methyltransferases, such as KMT2C (p.S3213L and p.K2797Rfs*26) (n = 4, 18%) or KMT2D (p.P2781T) (n = 1, 4.5%) were the most frequently mutated, followed by mutations of genes coding for chromodomain helicase DNA-binding proteins (n = 5, 23%) such as CHD2 (p.G870R, p.P1796S, and p.E1242K) (supplemental Figure 1B). These missense and frameshift indel mutations were predicted to be deleterious, leading to impairment of protein function. Besides, we found nonsense or missense mutations in other epigenetic genes previously described as frequently mutated in T- and B-cell lymphomas, such as TET2 (p.E149*), DNMT3A (p.Q678*), and CREBBP (p.L1825I).28-32
Other pathways involved in BI-ALCL
Genomic alterations in the PI3K signaling pathway were observed in 14% of cases and included point mutations of PTPN11 (n = 2) and PIK3CG (n = 1) genes. In 18% of cases, EOMES, known to be involved in lymphocyte development, showed deleterious in-frame insertion-deletion. One patient harbored a missense mutation in CARD11 (p.S621F), which is involved in TCR signaling. As to genes involved in cell-cycle control and apoptosis, 2 patients carried mutations in TP53 (p.R282W and p.C238Y) and 3 showed an in-frame deletion in TSC22D1 (p.Q509del) acting as a tumor suppressor through apoptosis induction. None of the samples showed germline mutation of TP53 (supplemental Figure 1B).
CNAs in BI-ALCL
All 22 samples were analyzed for CNA profiling, but CNA estimation from WES data was successful for 8 samples (2 in situ and 6 tumor-type BI-ALCL cases) but not evaluable in the remaining 14 samples, probably due to degradation of DNA from FFPE samples and/or the low proportion of neoplastic cells. The mean WES coverage was satisfactory in CNA-informative cases as well as in those with noninformative CNA (139× [113-192×] and 129× [83.5-153×], respectively). Four out of the 8 interpretable cases were polyploid (3 triploids and 1 tetraploid). All analyzed BI-ALCL genomes showed multiple aberrations, and most genomes were complex. CNA analysis showed more losses than gains (61% vs 39%). Recurrent CNAs were gains on chromosomes 2, 9p, 12p, and 21 and losses on 4q, 8p, 15, 16, and 20 (supplemental Figure 2A-B). The main gain region on chromosome 2 (:173929213-176957814) encompassed CDCA7, which is known to contribute to MYC-mediated tumorigenesis.33 Genes of the JAK/STAT pathway, including JAK1, STAT3, and STAT5B, were affected by CNAs, with concomitant gene mutations in some cases (detailed in supplemental Figure 2C). Several regions of loss and copy-neutral loss of heterozygosity encompassed JAK/STAT inhibitors and epigenetic regulators (supplemental Figure 2C). Loss of the 17p region harboring TP53 was detected in 3 cases, with concomitant TP53 mutation in 2 cases.
TDS
In order to validate and further expand our WES data, we performed TDS in 24 BI-ALCL cases, including 12 additional cases. The 12 double-screened cases showed a perfect concordance (Figure 1A). This extended BI-ALCL cohort confirmed the high rate of mutations targeting the JAK/STAT pathway, since at least 1 member of this pathway was mutated in 50% of the 12 additional cases. These recurrent mutations were observed in STAT3 (n = 3/12), JAK1 (n = 1/12), and STAT5B (n = 1/12), whereas 1 case harbored concurrent STAT3 and JAK1 mutations. Furthermore, 66% (n = 8/12) of the analyzed cases carried 1 or several deleterious mutations in epigenetic modifiers, including KMT2C (25%), CHD2 (17%), and CREBBP (17%).
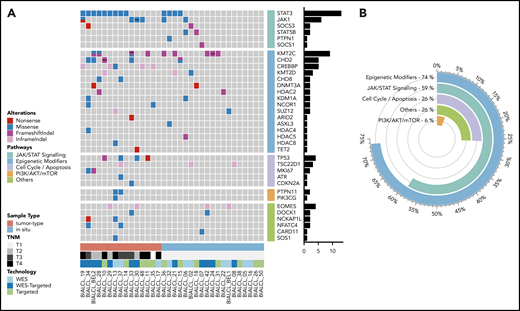
Mutational landscape of BI-ALCL. (A) Whole-exome sequencing and/or TDS in 34 BI-ALCLs. The heatmap of most significantly mutated genes is ordered according to biological pathways. (B) Functional pathways involved in BI-ALCL genomic abnormalities. The percentages refer to the frequency of alterations in the respective pathways.
Mutational landscape of BI-ALCL. (A) Whole-exome sequencing and/or TDS in 34 BI-ALCLs. The heatmap of most significantly mutated genes is ordered according to biological pathways. (B) Functional pathways involved in BI-ALCL genomic abnormalities. The percentages refer to the frequency of alterations in the respective pathways.
Altogether, 74% of the entire cohort of 34 BI-ALCLs sequenced by WES and/or TDS contained alterations of epigenetic regulators and histone modifiers, whereas mutations involving the JAK/STAT pathway were found in 59% of cases (Figure 1B). Somatic mutations in regulators of the cell cycle and apoptosis were less common (26%), including TP53 (12%) and PI3K/AKT (6%). Other altered pathways included lymphocyte development or TCR signaling (26%). A linear depiction of the protein domains of frequently mutated genes is shown in Figure 2 and supplemental Figure 3.
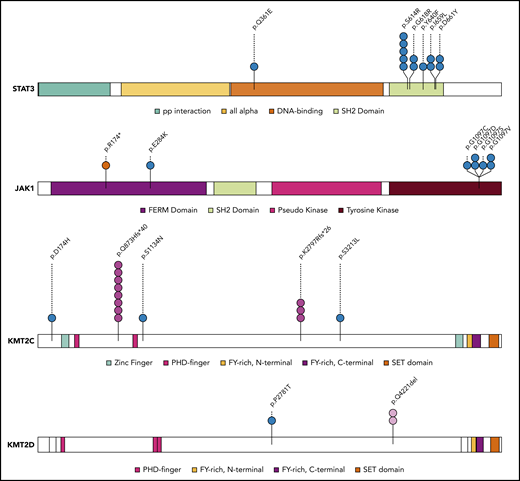
Mapping of protein variants produced by frequently mutated genes. Protein domains of STAT3, JAK1, KMT2C, and KMT2D were annotated according to the Pfam database. Each variant is represented by a colored circle, with missense mutations in blue, nonsense mutations in red, frameshift indel mutations in pink, and in-frame indels in light pink.
Mapping of protein variants produced by frequently mutated genes. Protein domains of STAT3, JAK1, KMT2C, and KMT2D were annotated according to the Pfam database. Each variant is represented by a colored circle, with missense mutations in blue, nonsense mutations in red, frameshift indel mutations in pink, and in-frame indels in light pink.
IHC
Using IHC, we investigated the consequences at the protein level of mutations targeting STAT3, STAT5B, KMT2C, and KMT2D (Figure 3). In this respect, 20 BI-ALCL cases with available material, including 15 STAT3 wild‐type and 5 STAT3-mutated cases, were analyzed using an anti-pSTAT3 antibody. All tested cases exhibited a strong nuclear positive staining indicating the presence of the phosphorylated form of STAT3 in lymphoma cells regardless of the mutational status (Figure 3A-B). Six cases, including the 2 STAT5B-mutated BI-ALCLs (cases 6 and 18), were also tested for phospho-STAT5 expression. The 2 STAT5B-mutated cases showed a strong nuclear expression of phospho-STAT5 in tumor cells, whereas the STAT5B wild-type cases were negative (supplemental Figure 4). In addition, as KMT2C and KMT2D take part in H3K4 trimethylation, staining using anti-H3K4me3 and H3K4me1 antibodies was performed in available KMT2C/D wild-type and mutated cases. As illustrated in Figure 3C-F, KMT2C/D wild-type BI-ALCLs showed H3K4me3 or H3K4me1 nuclear positivity in most of neoplastic cells (Figure 3C-D), whereas tumor cells of KMT2C/2D-mutated cases were negative for H3K4me1 and me3 antibodies or showed weak staining compared with reactive cells (Figure 3E-F). Automated quantification of the H3K4me1 and H3K4me3 staining further confirmed a significant difference between KMT2D/2C-mutated and KMT2D/2C wild-type cases, with a higher percentage of tumor cells negative and/or with a lower expression of H3K4me3 and H3K4me1 staining in mutated cases (n = 4 and n = 6, respectively) than unmutated ones (n = 8 and n = 4, respectively) (P < .0398 and P < .0086, respectively) (supplemental Figure 5).
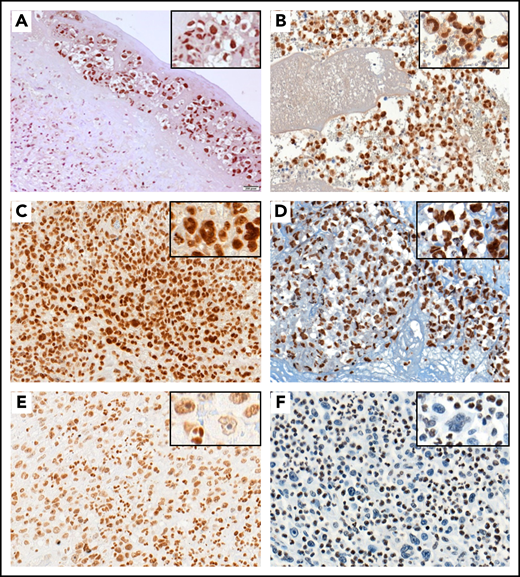
pSTAT3 and H3K4me3 immunostaining in BI-ALCL. Representative STAT3 wild-type (A) and STAT3-mutated (B) BI-ALCL cases showing nuclear expression of phospho-STAT3 in tumor cells regardless of the mutational status (A, hematoxylin and eosin stain, original magnification ×200; B, hematoxylin and eosin stain, original magnification ×200). Representative cases of KMT2C/2D wild-type BI-ALCLs showing strong nuclear expression of H3K4me3 (C, original magnification ×400) and H3K4me1 (D, original magnification ×400) with similar intensities in lymphoma cells and reactive surrounding cells. By contrast, the nuclear H3K4me3 staining in a representative case of KMT2C mutated BI-ALCL was weaker in tumoral cells than in reactive cells such as neutrophils (E, original magnification ×200). Similarly, nuclear H3K4me1 staining was lost in most neoplastic cells of a KMT2D-mutated BI-ALCL case (F, original magnification ×200).
pSTAT3 and H3K4me3 immunostaining in BI-ALCL. Representative STAT3 wild-type (A) and STAT3-mutated (B) BI-ALCL cases showing nuclear expression of phospho-STAT3 in tumor cells regardless of the mutational status (A, hematoxylin and eosin stain, original magnification ×200; B, hematoxylin and eosin stain, original magnification ×200). Representative cases of KMT2C/2D wild-type BI-ALCLs showing strong nuclear expression of H3K4me3 (C, original magnification ×400) and H3K4me1 (D, original magnification ×400) with similar intensities in lymphoma cells and reactive surrounding cells. By contrast, the nuclear H3K4me3 staining in a representative case of KMT2C mutated BI-ALCL was weaker in tumoral cells than in reactive cells such as neutrophils (E, original magnification ×200). Similarly, nuclear H3K4me1 staining was lost in most neoplastic cells of a KMT2D-mutated BI-ALCL case (F, original magnification ×200).
Correlations among genetic alterations, T-cell repertoire, tumor cell content, and pathological features
The Fisher exact test was used to compare the mutational profiles of the 2 BI-ALCL subtypes. Across all the samples, there was no significant difference of the total number of detected mutations and in the number of mutations involving the epigenetic modifiers between these 2 subtypes. However, tumor-type cases showed a higher rate of mutations in members of JAK/STAT pathway compared with in situ samples across all samples (80% vs 42% of cases; P = .038). In particular, STAT3 was significantly more mutated in tumor-type cases than in situ cases (n = 9/15 vs n = 4/19; P = .034). In addition, tumor-type cases were more often mutated in genes controlling the cell cycle than in situ cases (n = 7/15 vs n = 1/19, respectively; P = .025). A tendency for higher genomic complexity (ie, total number of breakpoints) in tumor-type than in situ samples (mean counts of 118 [82-194] and 76 [73-78], respectively) (P = .06) was seen. When samples were categorized according to the percentage of tumor cells (≤20%, 20% to 50%, and >50% of tumor cells per sample) determined by optical evaluation, there was no significant difference in terms of number of overall mutations as well as in mutations in the JAK/STAT pathway among the 3 categories. In an attempt to determine more precisely whether the number of alterations was related or not to tumor cell content, we compared genetic alterations (VAF of the dominant pathogenic mutation) with tumor cell content analyzed by the TCR repertoire using CapTCR-seq NGS. Overall, we found that the proportion of the dominant T-cell clone estimated by TCR sequencing was well correlated with the percentage of tumor cells evaluated by pathological examination (Spearman correlation s = 0.6) (supplemental Table 1). Considering the VAFs of the dominant pathogenic variant, the correlation with the dominant clonal TCRβ rearrangement was moderate (Spearman correlation s = 0.51). Indeed, whereas several cases (mostly tumor type [n = 7/9]) displayed high values of both the dominant pathogenic mutation and the dominant T-cell clone (VAF above half of the TCR value), others (mainly in situ BI-ALCLs [n = 6/11]) showed low VAF of the dominant mutation compared with the dominant T-cell clone (VAF below half of the TCR value), suggesting that the pathogenic mutation might belong to a subclone in these later cases (supplemental Table 1 and supplemental Figure 6).
Discussion
Chronic inflammation and subversion of cytokine receptors signaling was previously suspected to play a role in BI-ALCL pathogenesis. Our results strongly support this hypothesis, since ∼60% of our cases harbored somatic mutations in at least 1 member of the JAK/STAT cascade, among which STAT3 and JAK1 were the most frequently mutated genes. This is in keeping with the known STAT3 activation in BI-ALCL cell lines, with concomitant high levels of cytokines such as interleukin-6 (IL-6) and IL-10.34 We also observed constant nuclear expression of pSTAT3 in all BI-ALCL cases regardless of their mutational status. Since STAT3 mutations have been shown to increase the phosphorylation of STAT3 in response to IL-6,35 overexpression of cytokines and/or somatic mutations might be responsible for constitutive STAT3 activation in BI-ALCL.
This pathogenic mechanism appears not to be BI-ALCL specific, since STAT3 activation is known to occur in systemic ALCLs. JAK/STAT activation in BI-ALCL may be exacerbated, since the tumor occurs in an enclosed space in which cytokines can reach high levels due to chronic inflammation. Furthermore, 15% of our cases exhibited co-occurring mutations in 2 genes of the JAK/STAT pathway. Such double hits could potentialize JAK/STAT signaling in BI-ALCL. It was indeed previously demonstrated in vitro that double JAK1/STAT3 mutants could act synergistically to amplify STAT3 activation and sustain cell transformation.25 Likewise, the observed mutations in JAK/STAT-negative regulators and deletions in chromosomal regions containing these genes might further boost BI-ALCL growth.
In our series, 74% of cases showed deletions or inactivating mutations targeting KMT2C, CHD2, CREBBP, and KMT2D. The relevance of epigenetic alterations has been already emphasized in other T-cell lymphomas, such as those of T follicular helper origin; hepatosplenic T-cell lymphoma; ALCLs; intestinal T-cell lymphoma; peripheral T-cell lymphoma (PTCL), not otherwise specified; and extranodal natural killer/T-cell lymphoma.15,30,31,36-40 These latter malignancies, however, harbor mutations of the TET2, IDH2, DNMT3, and SETD2 genes, which were either absent or uncommon in our series (see supplemental Figure 7 and supplemental Table 4).
KMT2D and KMT2C are members of the mixed lineage leukemia methyltransferase family involved in the methylation of H3K4, an important process regulating gene transcription.41,42 So far, hundreds of KMT2C and KMT2D mutations have been identified, making them among the most frequently mutated genes in human cancers.41,42 Our results are in keeping with recent reports depicting alterations of these 2 genes in T-cell lymphomas like Sézary syndrome, extranodal natural killer/T-cell lymphoma, and PTCL, not otherwise specified.40,43-45 Loss of mixed lineage leukemia proteins function is known to affect cell differentiation and proliferation programs.46 Accordingly, we observed an absence or decrease of H3K4 methylation in KMT2C or KMT2D mutated BI-ALCL cases compared with wild-type cases. By contrast to the high frequency of missense mutation affecting the STAT3 gene, KMT2 genes were more often targeted by nonsense or frameshift mutations. This could be explained by the fact that STAT3 mutations (especially in its kinase domain) are known to induce oncogenic activation,25,47 whereas KMT2 mutations in the C-terminal portion (including the SET domain–containing lysine methyltransferase) are potentially inactivating, with subsequent loss of KMT2 protein reducing the methyltransferase activity.48,49 We could also detect frequent mutations in other genetic modifiers such as CHD2, CREBBP, and HDAC family members.
Altogether, we report here that the genomic profiling of BI-ALCL identifies common alterations in genes of the JAK-STAT pathway and in epigenetic modifiers. Most of the mutations observed in BI-ALCL have been reported in other PTCL subtypes,25,36,37,40,50-55 though at variable frequencies (supplemental Figure 7). However, the limited number of systemic or cutaneous ALCL cases with available sequencing data reported so far25,52 precludes definitive conclusion as to putative differences between the mutational landscape of BI-ALCL and other ALK-negative, systemic, or cutaneous ALCLs. Further larger studies are warranted to compare the genetic landscape of these different ALCL subsets.
TP53 germline mutations in BI-ALCL have been recently reported.14 We could not identify such mutations in our cases, although TP53 somatic mutations were present in 12% of cases and were associated with loss of the corresponding 17p region in 2 cases, whereas 17p loss was detected in another nonmutated case. These data suggest a significant impact of TP53 defect on BI-ALCL growth.
Five BI-ALCL cases studied by both WES and/or TDS did not show pathogenic mutations. For 2 samples with low tumor cell content, which also lacked TCR rearrangements, we cannot exclude a false-negative result. In the 3 remaining samples, we could ensure enrichment in tumor cell content by macrodissection and detect a monoclonal TCR rearrangement. The lack of mutations in these cases suggests that other mechanisms could contribute to STAT3 activation. This is consistent with our observation that all tested BI-ALCLs expressed pSTAT3, regardless to their mutational status. However, the possibility of minor subclones below the detection threshold of our sequencing approach cannot be totally ruled out.
Such an alternative mechanism is also supported by the finding that STAT3 is less frequently mutated in in situ cases than in tumor-type cases. This suggests a continuum of JAK/STAT pathway activation ranging from cytokine hypersecretion to JAK/STAT mutations. High cytokine levels could be solely responsible for some in situ lesions, whereas the aggressive tendency of tumor-type cases may be underlined by mutant JAK/STAT clones. In accordance with this hypothesis, TCR repertoire profile analysis indicated that in most tumor-type samples, the dominant pathogenic mutation was present in the majority of neoplastic T-cells.
To conclude, this genomic characterization of the largest BI-ALCL series reported to date not only confirms the key role of the JAK/STAT pathway but also highlights the importance of epigenetics in BI-ALCL pathogenesis. Compounds targeting these molecular alterations, which are already being explored in systemic ALCL and other T-cell lymphomas, might be considered in the future to treat the small proportion of BI-ALCL patients with aggressive refractory disease.
For original data, please contact Camille Laurent: laurent.c@chu-toulouse.fr.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Nathalie Van Acker (H3K4me1 staining), Eric Delabesse (TCR analysis), Giorgio Inghirami (expertise in ALCL sequenced data), Andrea Cerqueira Do Vale (technical support for TCR repertoire analysis), and François Lozach (probe design for TCR repertoire analysis). They acknowledge the experts from the Lymphopath network and the French registry on BI-ALCL, as well as the participants of the Tenomic consortium (a complete membership list appears in the supplemental Appendix).
This work was supported in part by institutional grants from the Institut National du Cancer (INCA, 2017-007), the Fondation pour la Recherche Médicale (FRM, Equipe Labellisée DEQ20160334875), the Leukemia Lymphoma Society (LLS SCOR 7013-17), the Laboratoire d’Excellence Toulouse Cancer (TOUCAN, contract ANR11-LABX), the Programme Hospitalo-Universitaire en Cancérologie CAPTOR (contract ANR11-PHUC0001), the Lymphome Study Association (LYSA), and the Institut Carnot Lymphome (CALYM).
Authorship
Contribution: Camille Laurent, A.N., V.F., N.A., P.B., L.X., and P.G. collected data; Camille Laurent, A.N., Cécile Laurent, L.X., and P.G. analyzed data and made the figures; Camille Laurent, A.N., Cécile Laurent, P.B., L.X., and P.G. designed the research and wrote the paper; Camille Laurent, A.N., Cécile Laurent, J.A., D.B., N.M., F.-X.F., L.L., A.B.-F., M.-H.D.-L., N.P., L.X., and P.G. performed experiments; Cécile Laurent, A.G., F.E., and B.T. provided expert statistic analysis; C.H., M.A., F.L.B., L.O., J.-M.S., F.R., R.B., and M.B. provided patient samples and clinical information; and Camille Laurent, A.N., L.X., P.B., P.G., A.T.-G., M.-P.C., L.M., A.-S.H., A.M., F.L., C.C.-C., and J.S. provided samples for analysis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Camille Laurent, Laboratoire d’Anatomie Pathologique, Institut Universitaire du Cancer de Toulouse-Oncopole, 1 Ave Irène Joliot-Curie, 31059 Toulouse, France; e-mail: laurent.c@chu-toulouse.fr; and Philippe Gaulard, Département de Pathologie, Groupe Hospitalier Henri Mondor, AP-HP, 94010 Créteil, France; e-mail: philippe.gaulard@aphp.fr.
