


Abstract
Venous thromboembolism (VTE), which includes deep vein thrombosis (DVT) and pulmonary embolism (PE), is the third most common cause of vascular death after heart attack and stroke. Anticoagulation therapy is the cornerstone of VTE treatment. Despite such therapy, up to 50% of patients with DVT develop postthrombotic syndrome, and up to 4% of patients with PE develop chronic thromboembolic pulmonary hypertension. Therefore, better therapies are needed. Although direct oral anticoagulants are more convenient and safer than warfarin for VTE treatment, bleeding remains the major side effect, particularly in cancer patients. Factor XII and factor XI have emerged as targets for new anticoagulants that may be safer. To reduce the complications of VTE, attenuation of thrombin activatable fibrinolysis inhibitor activity is under investigation in PE patients to enhance endogenous fibrinolysis, whereas blockade of leukocyte interaction with the vessel wall is being studied to reduce the inflammation that contributes to postthrombotic syndrome in DVT patients. Focusing on these novel antithrombotic strategies, this article explains why safer anticoagulants are needed, provides the rationale for factor XII and XI as targets for such agents, reviews the data on the factor XII– and factor XI–directed anticoagulants under development, describes novel therapies to enhance fibrinolysis and decrease inflammation in PE and DVT patients, respectively, and offers insights into the opportunities for these novel VTE therapies.
Introduction
Venous thromboembolism (VTE), which includes deep vein thrombosis (DVT) and pulmonary embolism (PE), occurs for the first time in 1 in 1000 individuals, and the incidence increases with age.1 Up to 12% of patients with PE and 6% of those with DVT die within 30 days.2 Of those who survive, 2% to 4% of patients with PE develop chronic thromboembolic pulmonary hypertension (CTEPH), which can be fatal, and 20% to 50% of patients with DVT develop postthrombotic syndrome (PTS), a chronic disorder characterized by leg pain and swelling that can lead to venous ulcers.3-5 Therefore, VTE is a common disorder associated with significant morbidity and mortality.
Anticoagulation is the mainstay of VTE treatment. Until recently, patients with acute VTE required an initial overlap of a rapidly acting parenteral anticoagulant, usually low-molecular-weight heparin (LMWH). with a vitamin K antagonist (VKA), such as warfarin.5 Although effective and safe, this regimen is inconvenient for patients and costly for health care systems because LMWH requires daily subcutaneous injection, and warfarin requires frequent monitoring and dose adjustments to ensure that the international normalized ratio is therapeutic.
Given in fixed doses without routine monitoring, direct oral anticoagulants (DOACs) have streamlined VTE treatment.6 The DOACs licensed for VTE treatment include dabigatran, which inhibits thrombin, and rivaroxaban, apixaban and edoxaban, which inhibit factor Xa (FXa).5 Their approvals were based on phase 3 trials demonstrating that DOACs were as effective as VKAs, but were associated with less bleeding.7 With similar efficacy, better safety, and greater convenience, guidelines give preference to DOACs over VKAs for VTE treatment in patients without active cancer,8 as well as an alternative to LMWH in selected patients with cancer-associated VTE.9
Although DOACs are safer than VKAs, bleeding remains the major side effect. This is particularly evident in cancer-associated VTE. Thus, although edoxaban and rivaroxaban were more effective than dalteparin for preventing recurrence in cancer patients, they were associated with more bleeding.10 Therefore, there is a need for safer anticoagulants, and FXII and FXI are being pursued as targets for the development of such agents.11
Advances in other VTE treatments have lagged compared with anticoagulation therapy. Other VTE treatments include systemic or catheter-directed thrombolytic therapy, as well as catheter-directed pharmacomechanical therapy and clot-extraction techniques.12-14 Thrombolytic therapy is reserved for patients with massive PEs or extensive proximal DVT. In patients with massive PEs, systemic thrombolytic therapy reduces mortality,15 decreases the risk of CTEPH, and improves quality of life.16,17 However, systemic thrombolytic therapy is of uncertain value in patients with submassive PEs, because any benefit in terms of reduction in PE-related morbidity and mortality is offset by an increase in major bleeding, including intracranial hemorrhage.18,19 Therefore, safer strategies are needed to enhance clot degradation in PE patients, thereby reducing mortality and the risk of CTEPH. Current efforts are targeting the regulators of fibrinolysis, including thrombin activatable fibrinolysis inhibitor (TAFI) and α2-antiplasmin.
In patients with DVT, PTS occurs despite anticoagulation therapy, and thrombolytic therapy does not appear to reduce the risk.12,13 Thus, compared with anticoagulation therapy alone, the use of catheter-directed pharmacomechanical therapy plus anticoagulation therapy did not reduce the risk of PTS in the Acute Venous Thrombosis Thrombus Removal with Adjunctive Catheter-Directed Thrombolysis trial.12 Consequently, new strategies for PTS prevention are needed.
In DVT, leukocytes and platelets tethered to the vein wall promote thrombus growth and inflammation by elaborating tissue factor and proinflammatory cytokines and by promoting coagulation.20,21 Leukocyte adhesion occurs when P-selectin glycoprotein ligand-1 (PSGL-1) on the surface binds P-selectin expressed on activated endothelial cells.22 Activated platelets also express P-selectin, which can bind them to leukocytes to form leukocyte-platelet aggregates that promote inflammation.22 Blocking the PSGL-1–P-selectin interaction attenuates thrombus formation and vein wall inflammation in animal models of DVT without causing bleeding,23 and studies are now underway to determine whether the same is true in humans.
Focusing on novel antithrombotic strategies for VTE, this article explains why safer anticoagulants for VTE treatment are needed, provides the rationale for FXII and FXI as targets for potentially safer anticoagulants, reviews available data on the FXII- and FXI-directed anticoagulants under development, describes novel therapies to enhance fibrinolysis in patients with PE and decrease inflammation in those with DVT, and offers insights into the opportunities for novel therapies for VTE.
Need for safer anticoagulants for VTE treatment
The goal of anticoagulation therapy is to attenuate thrombosis without perturbing hemostasis. Although DOACs come closer to this goal than VKAs, bleeding is not eliminated with DOACs. This is particularly evident in patients with cancer-associated VTE.10 Thus, in patients without active cancer, a meta-analysis revealed that DOACs are at least as effective as VKAs, but are associated with a 40% reduction in the risk of major bleeding.8 Different results were obtained when edoxaban or rivaroxaban was compared with dalteparin in patients with cancer-associated VTE in the Hokusai-VTE Cancer or the Anticoagulation Therapy in Selected Cancer Patients at Risk of Recurrence of Venous Thromboembolism (select-D) trials, respectively.24,25 A meta-analysis that focused on the 6-month outcomes in these trials reported a lower incidence of recurrent VTE with DOACs than with dalteparin (relative risk [RR], 0.65; 95% confidence interval [CI], 0.42-1.01) but a higher incidence of major bleeding (RR, 1.74; 95% CI, 1.05-2.88) and clinically relevant nonmajor bleeding (RR, 2.31; 95% CI, 0.85-6.28).10 The excess in major bleeding events with DOACs were mostly gastrointestinal bleeds that occurred in patients with gastrointestinal cancers, whereas most of the clinically relevant nonmajor bleeding events occurred in patients with gastrointestinal or urological cancers.24,25 Therefore, because of the increased risk of bleeding, DOACs can only be used in selected patients with VTE with active cancer, thereby highlighting the need for safer anticoagulants for this patient population.9
Rationale for targeting FXII and FXI
DOACs inhibit FXa or thrombin, downstream enzymes in the coagulation cascade. Interest has now shifted upstream to FXII and FXI. This shift stems from the fact that basic and epidemiological studies suggest that FXII and FXI are important in thrombosis, yet FXII has no role in hemostasis, and spontaneous bleeding is rare with congenital FXI deficiency. Therefore, FXII and FXI are important for thrombosis but less so for hemostasis. Consequently, FXII and FXI are promising targets for the development of safer anticoagulants.26-28
Several lines of evidence suggest that FXII and FXI are essential for thrombus stabilization and growth but less important for hemostasis. Thus, FXII- or FXI-deficient mice are protected from ischemic stroke, and clots that form after venous flow restriction in the inferior vena cava are smaller than those in wild-type mice.29-33 Likewise, FXI knockdown in baboons attenuates arteriovenous shunt thrombosis in a concentration-dependent manner once FXI levels decrease to <50% of normal.34 However, the extent of bleeding after tail tip amputation in mice deficient in FXII or FXI is indistinguishable from that in wild-type mice, consistent with the dispensable role of these coagulation factors in hemostasis.30,32,35 This dichotomy can be explained by the enhancement in thrombin generation that occurs when FXI is activated by FXIIa or thrombin, as well as by the subsequent augmented activation of TAFI, which attenuates fibrin clot degradation (Figure 1).36,37 Therefore, FXII and FXI are important for thrombosis but not for hemostasis.
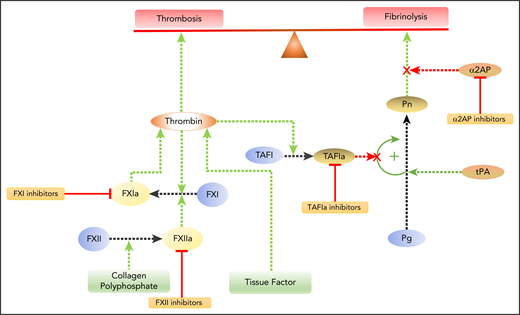
Modulating the balance between coagulation and fibrinolysis. Coagulation initiated by tissue factor exposed at sites of vascular injury results in thrombin generation. Thrombin converts fibrinogen to fibrin and activates platelets, which leads to thrombosis. Feedback activation of FXI by thrombin amplifies thrombin generation. FXI also can be activated via the intrinsic pathway when FXII is activated by collagen exposed at sites of denuding vascular injury or by polyphosphate released from activated platelets. Therefore, inhibitors of FXII or FXI have the potential to attenuate thrombosis. Plasmin (Pn) degrades fibrin to effect fibrinolysis. Plasminogen (Pg) is activated by tissue plasminogen activator (tPA). Fibrinolysis can be enhanced by inhibiting α2-antiplasmin (α2AP), the major inhibitor of Pn, or by inhibiting activated TAFIa. TAFIa attenuates fibrinolysis by releasing C-terminal lysine and arginine residues from degrading fibrin, thereby removing binding sites for Pg and tPA. TAFI is activated by thrombin after it binds to thrombomodulin on the cell surface, a process that links coagulation and fibrinolysis.
Modulating the balance between coagulation and fibrinolysis. Coagulation initiated by tissue factor exposed at sites of vascular injury results in thrombin generation. Thrombin converts fibrinogen to fibrin and activates platelets, which leads to thrombosis. Feedback activation of FXI by thrombin amplifies thrombin generation. FXI also can be activated via the intrinsic pathway when FXII is activated by collagen exposed at sites of denuding vascular injury or by polyphosphate released from activated platelets. Therefore, inhibitors of FXII or FXI have the potential to attenuate thrombosis. Plasmin (Pn) degrades fibrin to effect fibrinolysis. Plasminogen (Pg) is activated by tissue plasminogen activator (tPA). Fibrinolysis can be enhanced by inhibiting α2-antiplasmin (α2AP), the major inhibitor of Pn, or by inhibiting activated TAFIa. TAFIa attenuates fibrinolysis by releasing C-terminal lysine and arginine residues from degrading fibrin, thereby removing binding sites for Pg and tPA. TAFI is activated by thrombin after it binds to thrombomodulin on the cell surface, a process that links coagulation and fibrinolysis.
FXI has emerged as the preferred target for new anticoagulants. Its selection over FXII is based on several considerations. First, the epidemiological data supporting the role for FXI in thrombosis are stronger than those for FXII.38 Second, FXI appears to be a more important driver of thrombosis than FXII in nonhuman primates, because antibodies against FXI attenuate platelet and fibrin deposition more than those directed against FXII in the baboon arteriovenous shunt model.39 Third, when coagulation is initiated by tissue factor, back activation of FXI by thrombin has the potential to bypass FXII inhibition.40 Therefore, because of these considerations, strategies to inhibit FXI are more advanced than those targeting FXII.
Strategies to inhibit FXI and FXII
The strategies for FXI inhibition include antisense oligonucleotides (ASOs) that reduce hepatic synthesis of FXI,41 monoclonal antibodies that block FXI activation or FXIa activity,42,43 aptamers that bind FXI or FXIa,44 and small molecules that block the active site of FXIa or induce allosteric modulation (Table 1).45,46 Strategies to inhibit FXII focus on knock down of FXII with small interfering RNA molecules (siRNAs) or ASOs, aptamers that bind to FXII and/or FXIIa and block FXI activation, and monoclonal antibodies that inhibit FXIIa or bind to FXI and block its activation by FXIIa (Table 1).40,47-53 Development of potent and specific small molecule inhibitors is more difficult for FXIIa than for FXIa because FXIIa has an open active site cleft.54
Strategies to inhibit FXI or FXII
| Agents | Target | |
|---|---|---|
| FXI | FXII | |
| ASOs | Reduce hepatic synthesis of FXI by inducing catalytic degradation of FXI mRNA (eg, IONIS-416858)*,† | Reduce hepatic synthesis of FXII by inducing catalytic degradation of FXII mRNA |
| Monoclonal antibodies | Suppress FXIa generation and/or inhibit FXIa activity (eg, BAY1213790 (osocimab),*,† BAY1831865, MAA868, AB023*) | Bind to FXII and block its activation (eg, 9A2, 15H8, AB052) |
| Bind to FXIIa and blocks its activity (eg, CSL312,* 3F7) | ||
| Small peptidomimetic or peptide inhibitors | Bind to catalytic domain (eg, BMS-986177,*,† EP-7041, ONO-5450598) | Bind to catalytic domain (eg, rHA-infestin 4, FXII618, 3-carboxamide coumarins) |
| Aptamers | Bind to FXI and/or FXIa and block its activity (eg, 11.16, 12.7) | Bind to FXII and/or FXIIa and block FXI activation (eg, R4cXII-1) |
| Agents | Target | |
|---|---|---|
| FXI | FXII | |
| ASOs | Reduce hepatic synthesis of FXI by inducing catalytic degradation of FXI mRNA (eg, IONIS-416858)*,† | Reduce hepatic synthesis of FXII by inducing catalytic degradation of FXII mRNA |
| Monoclonal antibodies | Suppress FXIa generation and/or inhibit FXIa activity (eg, BAY1213790 (osocimab),*,† BAY1831865, MAA868, AB023*) | Bind to FXII and block its activation (eg, 9A2, 15H8, AB052) |
| Bind to FXIIa and blocks its activity (eg, CSL312,* 3F7) | ||
| Small peptidomimetic or peptide inhibitors | Bind to catalytic domain (eg, BMS-986177,*,† EP-7041, ONO-5450598) | Bind to catalytic domain (eg, rHA-infestin 4, FXII618, 3-carboxamide coumarins) |
| Aptamers | Bind to FXI and/or FXIa and block its activity (eg, 11.16, 12.7) | Bind to FXII and/or FXIIa and block FXI activation (eg, R4cXII-1) |
mRNA, messenger RNA.
Under evaluation in phase 2 clinical trials.
Discussed in text.
The mode of administration of FXI or FXII inhibitors differs depending on the agent. siRNAs, ASOs, antibodies, and aptamers require parenteral administration, whereas small molecule active site inhibitors have the potential for parenteral or oral delivery. It takes up to 4 weeks for siRNA or ASO treatment to lower FXII or FXI levels into the therapeutic range, which limits their utility for acute therapy. In contrast, antibodies, aptamers, and small molecules have a rapid onset of action. The long half-life of FXI-directed antibodies or the FXI ASO could be problematic if there is bleeding with trauma or surgery. Although the anticoagulant effect of the ASO can readily be overcome with FXI replacement, this will not be the case for antibodies. Instead, reversal with anti-idiotype antibodies or bypass with low-dose recombinant FVIIa may be needed. Bleeding complications are less likely to be a concern with long-acting FXII-directed strategies because FXII is dispensable for hemostasis. Therefore, each strategy has strengths and weaknesses for clinical development.
Clinical trials with FXI and FXII inhibitors
No phase 2 study with FXII inhibitors is currently underway. In contrast, phase 2 studies with FXI inhibitors include those with IONIS-416858, BAY 1213790 (now known as osocimab), and BMS986177.41,42,45,55 Each agent will be discussed separately.
IONIS-416858
The first FXI-directed anticoagulant to be tested in humans was IONIS-416858, the FXI-directed ASO, which is given subcutaneously (Table 1).41 In healthy volunteers, IONIS-416858 reduced FXI antigen and activity levels in a dose-dependent manner. A phase 2 study in patients undergoing elective knee replacement randomized 300 patients to receive subcutaneous IONIS-416858 (at doses of 200 or 300 mg starting 35 days prior to surgery) or enoxaparin (40 mg once daily, with the first dose given 12 hours before or after surgery).41 Treatments were continued for ≥10 days, at which point bilateral venography was performed. The primary efficacy outcome was VTE, which included asymptomatic DVT detected by venography, objectively confirmed symptomatic DVT or PE, and VTE-related mortality. The principal safety outcome was the composite of major and clinically relevant nonmajor bleeding. The primary efficacy outcome occurred in 36 of 134 patients (27%) and in 3 of 71 patients (4%) who received the 200- and 300-mg doses of IONIS-416858, respectively, compared with 21 of 69 patients (30%) who received enoxaparin.26 The 200-mg IONIS-416858 regimen was noninferior to enoxaparin, whereas the 300 mg ASO regimen was superior (P < .001). The rates of major and clinically relevant nonmajor bleeding were 3% in both IONIS-416858 groups and 8% in the enoxaparin group; differences were not statistically significant. Therefore, lowering FXI levels reduces postoperative VTE more than enoxaparin without significantly increasing the risk of bleeding.
Two phase 2 studies are evaluating the safety of IONIS-416858 in patients on chronic hemodialysis (NCT02553889 and NCT03358030).56,57 Despite the use of heparin during dialysis, preliminary data suggest that, compared with placebo, lowering FXI levels in such patients reduces clotting in the air trap and on the dialysis membrane.58
Osocimab
Osocimab is a fully human immunoglobulin G1 antibody that inhibits FXIa. In a single ascending-dose phase 1 study, osocimab demonstrated favorable safety and tolerability after IV infusion; there were no cases of bleeding or clinically relevant antidrug antibody formation (Table 1).42 Of the 83 volunteers, 1 (1.2%) had an infusion reaction. Osocimab reduced FXI activity and prolonged the activated partial thromboplastin time and the whole blood clotting time (as determined using rotational thromboelastometry) in a dose-dependent manner. It had no effect on bleeding times.
The efficacy and safety of osocimab have been compared with those of enoxaparin and apixaban in the Factor XIa Inhibition for the Prevention of Venous Thromboembolism in Patients Undergoing Total Knee Arthroplasty (NCT03276143) trial.55 The study was conducted in 2 parts. In the first part, osocimab was administered postoperatively, whereas in the second part, 2 doses of osocimab selected from part 1 were evaluated with preoperative administration. As per their labels, enoxaparin and apixaban were started postoperatively, although a preoperative dose of enoxaparin was allowed. The study has been completed but the results have not yet been disclosed.
BMS986177
An oral FXIa inhibitor, BMS986177 (which is now designated as JNJ-70033093) has undergone single and multiple ascending-dose phase 1 evaluation in healthy volunteers with or without concomitant aspirin (Table 1).45,59,60 In addition, a single dose of BMS986177 was evaluated in 6 patients with end stage renal disease who were on hemodialysis. Therefore, BMS986177 has undergone robust phase 1 evaluation, but the results have not been published.
The ongoing phase 2 Antithrombotic treatment with Factor XIa inhibition to Optimize Management of Acute Thromboembolic Events study (NCT03766581) is comparing BMS986177 (25, 50, 100, or 200 mg once or twice daily) with placebo in patients with high-risk transient ischemic attack or small ischemic stroke.61 All patients will be treated with aspirin plus clopidogrel for 30 days, followed by aspirin alone thereafter. Evidence of new stroke or intracranial bleeding on repeated brain imaging will be the primary efficacy and safety outcome, respectively. A second phase 2 study will compare similar doses of BMS-986177 with enoxaparin in patients undergoing elective knee replacement surgery (NCT03891524).45
New treatments for PE
About 20% of patients with PE have submassive PE, which is defined as PE without hypotension (systolic blood pressure < 90 mm Hg) but with right ventricular enlargement or dysfunction and/or elevated biomarkers, such as troponin or brain natriuretic peptide.62 Up to 5% of such patients die, and their risk of CTEPH is likely higher than that in patients with less extensive PE. Full-dose or half-dose thrombolytic therapy increases the risk of major bleeding, including intracranial bleeding, in patients with submassive PE; neither regimen has been shown to reduce mortality.63,64 Although catheter-directed thrombolysis using very low doses of thrombolytic agents, with or without concomitant ultrasound or clot-extraction techniques, may be safer than systemic thrombolytic therapy, these procedures are expensive, and data supporting their use are limited.14 Therefore, simple and safe strategies to enhance fibrinolysis in patients with submassive PE are needed.
Inhibitors of activated TAFI (TAFIa) are being investigated for enhancement of endogenous fibrinolysis (Table 2).65 When thrombin is generated, TAFI is activated by the thrombin-thrombomodulin complex. TAFIa attenuates fibrinolysis by removing C-terminal lysine and arginine residues from partially degraded fibrin.66 These residues augment fibrinolysis by providing additional binding sites for plasminogen and tissue plasminogen activator during the clot-degradation process. Therefore, inhibition of TAFIa enhances fibrinolysis by preventing the removal of these lysine and arginine residues.
Strategies to enhance fibrinolysis
| Agents | Target | |
|---|---|---|
| TAFIa | α2-Antiplasmin | |
| Monoclonal antibodies | Bind to TAFI and block its activation (eg, MA-T9H11, MA-RT30D8, MA-TCK11A9, MA-TCK26D6, MA-T12D11, mAbTAFI/TM#16, MA-TCK27A4) | Bind to circulating and fibrin-bound α2-antiplasmin and neutralize its activity [eg, DS-9231 (TS-23)] |
| Small molecule inhibitors | Bind to TAFIa and block its activity (eg, DS-1040,* S62798) | Not reported |
| Agents | Target | |
|---|---|---|
| TAFIa | α2-Antiplasmin | |
| Monoclonal antibodies | Bind to TAFI and block its activation (eg, MA-T9H11, MA-RT30D8, MA-TCK11A9, MA-TCK26D6, MA-T12D11, mAbTAFI/TM#16, MA-TCK27A4) | Bind to circulating and fibrin-bound α2-antiplasmin and neutralize its activity [eg, DS-9231 (TS-23)] |
| Small molecule inhibitors | Bind to TAFIa and block its activity (eg, DS-1040,* S62798) | Not reported |
Under evaluation in phase 2 clinical trials; discussed in text.
DS-1040 is a potent small molecule inhibitor of TAFIa. In a phase 1 single ascending-dose study, 103 young (18-45 years of age) or elderly (65-75 years of age) subjects were randomized to DS-1040 (in doses ranging from 0.1 mg to 40 mg) or to placebo, which was administered as 30-minute or 24-hour IV infusions (Table 2).67 All doses of DS-1040 were well tolerated, and the bleeding time remained within the normal range. DS-1040 decreased TAFIa activity and clot lysis times in a dose- and time-dependent manner. Therefore, DS-1040 appears to be safe.
An ongoing phase 2 study is comparing an increasing dose of DS-1040 with placebo in anticoagulated patients with submassive PE (NCT02923115).68 The primary outcome is clinically relevant bleeding, but the effect of DS-1040 on thrombus volume is also being evaluated by computed tomography pulmonary angiography. If DS-1040 enhances fibrinolysis, thrombus volume in the pulmonary arteries 48 hours and 30 days after its administration should be less than that observed with placebo. Such findings would support further development of DS-1040, with the expectation that it may reduce mortality and decrease the risk of CTEPH in patients with extensive PE.
A second target for enhancement of endogenous fibrinolysis is α2-antiplasmin, which inhibits plasmin, the enzyme that degrades fibrin. In addition to circulating inhibitor, α2-antiplasmin cross-linked onto fibrin renders thrombi resistant to premature lysis.69,70 Consequently, inhibition of α2-antiplasmin has the potential to promote clot lysis. The efficacy and safety of this approach remain to be defined.
Monoclonal antibodies against α2-antiplasmin have been synthesized71 and show promise in animal models.72 TS-23 (also known as DS-9231), an α2-antiplasmin inactivating antibody, was well tolerated in a first-in-human study and suppressed α2-antiplasmin activity and increased D-dimer levels in a dose-dependent manner. There were no bleeding complications. Despite these promising data, TS-23 has not yet been evaluated in patients with PE (Table 2).73
New treatments for DVT
PTS is a common complication of DVT that reduces quality of life.74 More than one third of DVT patients develop PTS, and of these, up to 10% develop severe PTS, which can be associated with venous ulceration. Although the symptoms of PTS vary, they include aching pain, heaviness and swelling in the affected limb. Typically, the symptoms are aggravated by standing or walking and are relieved with rest and leg elevation. The signs of PTS include limb swelling, as well as telangiectasia and brown discoloration around the ankles and calves. In severe cases there may be tender thickening of the skin, so-called “lipodermatosclerosis,” and venous ulcers that are painful and slow to heal. Therefore, PTS is burdensome for patients, and its management is costly for health care systems.
PTS occurs because of venous hypertension secondary to persistent venous obstruction and venous valve reflux. Persistent venous obstruction is the more important mechanism, because ultrasound evidence of residual venous obstruction 6 months after DVT, with or without associated venous reflux, predicts the development of PTS, whereas venous reflux alone does not.75,76 Up to 50% of patients with DVT have persistent venous obstruction despite anticoagulation therapy, which places them at risk for PTS. Neither graded compression stockings nor catheter-directed pharmacomechanical therapy has been proven to reduce the risk of PTS.12,77 Therefore, new therapeutic strategies are needed.
Vein wall inflammation is a hallmark of PTS. In animal models of DVT, leukocytes and platelets adhere to the vein wall.78 When endothelial cells are activated by thrombin, P-selectin stored in Weibel-Palade bodies is translocated to the cell surface. E-selectin expression on endothelial cells comes later in response to inflammatory cytokine stimulation. P- and E-selectin tether leukocytes rolling on the vein wall by binding to PSGL-1 on the leukocyte surface (Figure 2).79 Adherent leukocytes become activated and express tissue factor and proinflammatory cytokines that promote clotting and inflammation.21 In animal models of DVT, inhibition of PSGL-1–selectin interactions attenuates thrombosis to a similar extent as enoxaparin in some studies.23,80,81 However, unlike enoxaparin, blockade of PSGL-1–selectin interactions does not increase bleeding.23,80,81 Therefore, by decreasing thrombosis and vein wall inflammation, inhibitors of the PSGL-1–P-selectin interaction may attenuate PTS in patients with DVT.

Modulating the cellular inflammatory response to prevent thrombosis and PTS. Most venous thrombi originate in the valve pockets when endothelial cells activated by stasis and resultant hypoxia express adhesion molecules, such as P- and E-selectin, on their surface. Leukocytes express PSGL-1 on their surface, which tethers them to the P- or E-selectin on activated endothelial cells. Tethered leukocytes express tissue factor, which initiates coagulation, and release proinflammatory cytokines. Together, these factors promote thrombosis and inflammation, which can lead to PTS. SelK2 and GMI-1271 have the potential to block this process by inhibiting PSGL-1 and E-selectin, respectively.
Modulating the cellular inflammatory response to prevent thrombosis and PTS. Most venous thrombi originate in the valve pockets when endothelial cells activated by stasis and resultant hypoxia express adhesion molecules, such as P- and E-selectin, on their surface. Leukocytes express PSGL-1 on their surface, which tethers them to the P- or E-selectin on activated endothelial cells. Tethered leukocytes express tissue factor, which initiates coagulation, and release proinflammatory cytokines. Together, these factors promote thrombosis and inflammation, which can lead to PTS. SelK2 and GMI-1271 have the potential to block this process by inhibiting PSGL-1 and E-selectin, respectively.
SelK2 is a humanized monoclonal antibody that binds PSGL-1 with high affinity and specificity and blocks its ability to interact with selectins and chemokines (Figure 2). The ongoing COURSE study (NCT03812328) is comparing the safety and efficacy of SelK2 alone, enoxaparin alone, or SelK2 plus enoxaparin in patients undergoing elective knee replacement surgery.82 This study will determine the antithrombotic and anti-inflammatory effects of SelK2.
GMI-1271 is an E-selectin antagonist that attenuates thrombosis and inflammatory markers in animal models without increasing bleeding (Figure 2).81,83 In 2 phase 1 studies in healthy volunteers, GMI-1271 was well tolerated and did not prolong clotting times.84,85 GMI-1271 is now being compared with placebo for the prevention of thrombus extension in patients with calf DVT (NCT02744833).86
Conclusions and future directions
VTE is the third most common cause of vascular death after heart attack and stroke. Although DOACs have simplified the prevention and treatment of VTE, they have not reduced the risk of CTEPH or PTS. Strategies that enhance endogenous fibrinolysis or attenuate the thrombosis-inflammation axis may address these problems when either is used alone or in combination with an anticoagulant. Therefore, the results of ongoing studies with TAFIa inhibitors, SelK2 and GMI-1271, are eagerly awaited.
Anticoagulants that are safer than DOACs are needed for the treatment of VTE in cancer patients. Additional clinical trials are needed to determine whether FXII or FXI inhibitors will fill this niche. FXII or FXI inhibitors may also fill other gaps. For example, like warfarin, DOACs are contraindicated in pregnancy because they cross the placenta; in contrast, ASOs do not.87 Therefore, with a once-monthly injection, ASOs may be safe and more convenient than LMWH for the prevention or treatment of thrombosis in pregnancy.
DOACs also are contraindicated in patients with mechanical heart valves because dabigatran was less effective than warfarin at preventing ischemic stroke in such patients.88 Mechanical heart valves and catheters trigger clotting by activating FXII.48,89,90 Consequently, DOACs may also be less effective than LMWH for prevention or treatment of central venous catheter thrombosis. FXII or FXI inhibitors may be more effective than DOACs in this setting because they block the root cause of catheter thrombosis. In support of this concept, catheters coated with corn trypsin inhibitor, a potent and specific inhibitor of FXIIa, remain patent longer than do uncoated catheters when inserted in the jugular veins of rabbits,90 and FXII or FXI knockdown with ASOs prolonged the time to occlusion of such catheters by more than twofold.48 Therefore, catheter thrombosis may be another problem that is best addressed with inhibitors of FXII or FXI.
In summary, advances in anticoagulation have simplified VTE prevention and treatment. The next frontier is development of safer anticoagulants for VTE treatment in cancer patients and the introduction of novel strategies to reduce the risk of CTEPH and PTS. Progress has been made on all of these fronts, and the success of these ventures will unfold over the next few years.
Acknowledgments
The authors thank Jim Fredenburgh for assistance with creating Figure 2.
This work was funded by grants from the Canadian Institutes of Health Research and the Heart and Stroke Foundation of Canada. J.I.W. holds the Canada Research Chair in Thrombosis and the Heart and Stroke Foundation J. F. Mustard Chair in Cardiovascular Research. N.C.C. is the recipient of a McMaster University, Department of Medicine Internal Research Career Award.
Authorship
Contribution: J.I.W. and N.C.C. developed the initial outline of the article; J.I.W. wrote the first draft; and both authors provided critical reviews and edits to produce the final manuscript.
Conflict-of-interest disclosure: J.I.W. has served as a consultant and has received honoraria from Bayer, Janssen, Johnson & Johnson, Boehringer Ingelheim, Bristol-Myers Squibb, Pfizer, Daiichi Sankyo, Ionis, Anthos, and Tetherex. N.C.C. has received speaker fees from Bayer for activities outside the scope of this work.
Correspondence: Jeffrey I. Weitz, Thrombosis and Atherosclerosis Research Institute, 237 Barton St E, Hamilton, ON L8L 2X2, Canada; e-mail: weitzj@taari.ca.
