

Key Points
LBCL with IRF4 rearrangement displays a mutational profile distinct from other LBCLs affecting pediatric and young adult patients.
Age, high genetic complexity, ABC profile, and TP53 mutations are associated with poor prognosis in pediatric and young adult LBCL.

Abstract
Pediatric large B-cell lymphomas (LBCLs) share morphological and phenotypic features with adult types but have better prognosis. The higher frequency of some subtypes such as LBCL with IRF4 rearrangement (LBCL-IRF4) in children suggests that some age-related biological differences may exist. To characterize the genetic and molecular heterogeneity of these tumors, we studied 31 diffuse LBCLs (DLBCLs), not otherwise specified (NOS); 20 LBCL-IRF4 cases; and 12 cases of high-grade B-cell lymphoma (HGBCL), NOS in patients ≤25 years using an integrated approach, including targeted gene sequencing, copy-number arrays, and gene expression profiling. Each subgroup displayed different molecular profiles. LBCL-IRF4 had frequent mutations in IRF4 and NF-κB pathway genes (CARD11, CD79B, and MYD88), losses of 17p13 and gains of chromosome 7, 11q12.3-q25, whereas DLBCL, NOS was predominantly of germinal center B-cell (GCB) subtype and carried gene mutations similar to the adult counterpart (eg, SOCS1 and KMT2D), gains of 2p16/REL, and losses of 19p13/CD70. A subset of HGBCL, NOS displayed recurrent alterations of Burkitt lymphoma–related genes such as MYC, ID3, and DDX3X and homozygous deletions of 9p21/CDKN2A, whereas other cases were genetically closer to GCB DLBCL. Factors related to unfavorable outcome were age >18 years; activated B-cell (ABC) DLBCL profile, HGBCL, NOS, high genetic complexity, 1q21-q44 gains, 2p16/REL gains/amplifications, 19p13/CD70 homozygous deletions, and TP53 and MYC mutations. In conclusion, these findings further unravel the molecular heterogeneity of pediatric and young adult LBCL, improve the classification of this group of tumors, and provide new parameters for risk stratification.
Introduction
Large B-cell lymphomas (LBCLs) in children and young adults have morphological and phenotypic features similar to those observed in their adult counterparts. However, the more favorable outcome of most pediatric patients after high-dose chemotherapy may be due, among other factors, to a different underlying biology.1 Recent molecular studies of diffuse LBCLs (DLBCLs) not otherwise specified (NOS) in adults revealed that the heterogeneity of these tumors is mainly related to cell-of-origin (COO) subtyping into germinal center B cells (GCBs) or activated B cells (ABCs), and a plethora of genomic alterations defining specific clusters with different clinical manifestations and outcome.2-7
Aggressive mature B-cell lymphomas in children and young adults include Burkitt lymphoma (BL), primary mediastinal large B-cell lymphoma (PMBL), and DLBCL, NOS. The first 2 subtypes have been extensively studied with now well-established profiles of genomic alterations.8-12 However, the molecular characterization of DLBCL, NOS in this age group is less defined. In fact, the constellation of LBCL in these patients seems more diverse than initially recognized. Clinicopathologic studies of LBCL in children have identified 2 additional tumors subtypes, included in the recent update of the World Health Organization (WHO) classification as provisional entities, that have overlapping features with BL and DLBCL.13 Burkitt-like lymphoma with 11q aberration (BLL-11q) is a high-grade B-cell lymphoma (HGBCL) that was initially considered BL related but without MYC translocations.14 However, 2 recent molecular studies have identified that these tumors lack the common BL mutations in the TCF3-ID3 axis and carry alterations closer to those found in GCB-DLBCL, although with differences suggesting they are a specific DLBCL subtype.15,16 LBCL with IRF4 rearrangement (LBCL-IRF4) predominates in pediatric population, has a favorable outcome after therapy, and consistently expresses IRF4 due to translocation.17-20 These cases display a complex pattern of chromosomal changes, but their mutational profile and possible relationship to other LBCLs is not known.21 The last WHO classification has also recognized the category of HGBCL that encompasses a spectrum of morphological appearances from blastoid to cases with intermediate features between BL and DLBCL.13 The mutational profile of these tumors is not well known, but some studies in adults have identified the simultaneous presence of characteristic mutations of both BL and DLBCL.22,23 The genomic features of these tumors in pediatric populations and their relationship to other LBCLs in this group of patients are not known.
Pediatric LBCLs and BL are treated using the same therapeutic protocols.1,24 Although generally curable with this intensive chemotherapy, ∼10% of cases relapse.1 Biological prognostic parameters predicting an adverse outcome have been extensively studied in adult DLBCL5,6,25,26 but are less well defined in pediatric and young adult tumors, with only few studies reported.24,27 A better understanding of the molecular pathogenesis of these tumors may assist in defining management protocols better suited to the biology of the disease. In the present study, we aimed to extensively characterize the molecular landscape of LBCL in the pediatric and young adult population and identify clinically relevant molecular features specific to different subtypes that are distinct from adult cases.
Methods
Patients and samples
Sixty-three patients <26 years with LBCL were included in the study and centrally reviewed by 3 hematopathologists (B.G.-F., O.B., and E.C.). Cases were classified according to WHO criteria13 into DLBCL, NOS (n = 31); LBCL-IRF4 (n = 20); and HGBCL, NOS (n = 12). No HGBCLs with MYC and BCL2/BCL6 rearrangements were identified. Fifty-three cases (51 primary and 2 relapses obtained 10 and 23 months after first diagnosis) were gathered in a centralized review supported by Sociedad Española de Hematología y Oncología Pediátrica or from the hematopathology files of Hospital Clínic of Barcelona, Spain. Additionally, 9 LBCL-IRF4 and 1 DLBCL, NOS were consultation cases from the University of Tübingen (Tübingen, Germany), National Institutes of Health (Bethesda, MD), and Children’s Hospital Los Angeles (Los Angeles, CA). Moreover, samples at relapse from 3 patients with primary tumor available were investigated. BL, BLL-11q, and PMBL cases were excluded. This study was approved by our institutional review board and in accordance with the Declaration of Helsinki.
Immunohistochemistry and FISH
Immunohistochemistry and fluorescence in situ hybridization (FISH) analyses were performed using standard protocols. The morphology, growth pattern, cytology, and immunohistochemical stains together with Epstein-Barr virus (EBV) in situ hybridization were evaluated as part of the diagnostic workup (supplemental Table 1, available on the Blood Web site). Cases were classified as germinal center (GC) and non-GC subtypes according to the Hans algorithm.28 Genetic alterations of BCL2, BCL6, MYC, IRF4, CIITA, and IGH were analyzed by commercial (Metasystems, Altlußheim, Germany) or homemade FISH probes.17,29
Targeted NGS and mutational analysis
Fifty-five LBCLs from 52 patients were examined for the mutational status of 96 B-cell lymphoma–related genes (supplemental Table 2) using the SureSelectXT Target Enrichment System Capture next-generation sequencing (NGS) strategy library (Agilent Technologies, Santa Clara, CA) before sequencing on MiSeq equipment (Illumina, San Diego, CA) (supplemental Methods; supplemental Figures 1 and 2). The contribution of previously defined mutational signatures was calculated for each gene (supplemental Methods). Variant verification was performed using the Ampliseq NGS method (Illumina) (supplemental Table 3) and/or by Sanger sequencing analysis using the primers detailed in supplemental Table 4. The previously published mutational profile of 144 adult DLBCLs was used for comparisons.26
DNA CN alteration analysis
Copy-number (CN) alterations were examined in 59 LBCLs from 55 patients using Oncoscan or single-nucleotide polymorphism array platforms (Thermo Fisher Scientific, Waltham, MA) according to standard protocols (supplemental Methods). Gains and losses and CN neutral loss of heterozygosity (CNN-LOH) regions were evaluated using Nexus Biodiscovery v9.0 software (Biodiscovery, Hawthorne, CA). Additional previously published CN data were used for comparison.26,30
Gene expression profile by NanoString
COO classification was performed using Lymph2Cx assay (NanoString, Seattle, WA).31 The Lymph3Cx assay was used for detection of molecular PMBL (mPMBL).32 The NanoString PanCancer Immune Profiling Panel was also used to investigate additional gene expression differences between different subsets of LBCL (supplemental Methods).
Statistical methods
Survival probabilities were estimated with the Kaplan-Meier method and differences assessed by the log-rank test. Event-free survival (EFS) was calculated as previously described.33 Differences in the distribution of individual parameters among patient subsets were analyzed by Fisher’s exact test for categorized variables, and the Student t test for continuous variables. Nonparametric tests were applied when necessary. Only mutations and genomic aberrations present in 5% of the cases and affecting a minimum of 4 cases were accounted for comparisons. The P values for multiple comparisons were adjusted using the Benjamini-Hochberg correction (false discovery rate). A cutoff of P = .05 was considered significant unless otherwise indicated. Statistical analyses were carried out using R software v3.5.0.
Results
Clinicopathological characteristics
Twenty cases were classified as LBCL-IRF4 (11 females, 9 males; median age, 14 years; range, 5-22 years). Eight patients had nodal involvement, mainly in the head and neck region, and 8 had tonsillar disease (Table 1). The other 4 patients (20%) had primary extranodal presentation in the gastrointestinal tract (2 cases), liver, and larynx. Histologically, 9 cases were purely diffuse, 5 cases showed a nodular growth pattern, and 6 displayed both follicular and diffuse areas. All cases showed positivity for MUM1/IRF4 and BCL6, whereas CD10 and BCL2 were positive in 11 and 10 cases, respectively (Figure 1). Five out of 12 cases coexpressed CD5, and all cases analyzed were negative for EBV. FISH studies identified the IRF4 translocation in 17 out of the 19 investigated cases, and the remaining 2 negative cases had breaks of the IGH locus (Figure 2). None of the 11 LBCL-IRF4 cases interrogated carried BCL6 or BCL2 rearrangements. The Lymph2Cx assay predicted 10 cases (72%) as GCB, 3 (21%) as unclassified, and only 1 (7%) as ABC. The IRF4/MUM1 messenger RNA levels detected by this assay were significantly higher than in DLBCL, NOS and HGBCL, NOS (P < .01) (supplemental Figure 3).
Pathological and clinical features of 20 LBCL cases with IRF4 rearrangement
| Case | Age (y), gender | Biopsy site | Growth pattern | Immunohistochemistry | FISH | COO NanoString (Lymph2Cx) | Stage* | Treatment | Outcome, follow-up | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD10 | BCL6 | BCL2 | IRF4/MUM1 | IRF4 | IGH | ||||||||
| D1 | 5, F | LN | Follicular and diffuse | + | + | + | + | ||||||
| D2 | 14, M | LN | Follicular | + | + | − | + | R | GCB | Surgical excision | |||
| D15 | 5, F | Tonsil | Diffuse | − | + | + | + | R | R | GCB | II | CT-P | CR, 49 mo |
| D16 | 14, F | Tonsil | Diffuse | − | + | − | + | R | II | CT-P | CR, 99 mo | ||
| D17 | 21, M | Liver | Diffuse | − | + | + | + | R | ABC | I-E | CT-A | CR, 10 mo † | |
| D20 | 17, M | Ileum | Diffuse | + | + | + | + | N | R | UNC | II | CT-P | CR, 72 mo |
| D21 | 8, M | Tonsil | Follicular | + | + | + | + | R | I | CT-P | CR, 36 mo | ||
| D23 | 21, F | Inguinal LN | Diffuse | − | + | + | R | R | GCB | IV-A | CT-A | CR, 40 mo | |
| D31 | 12, F | Cervical LN | Diffuse | − | + | + | + | N | R | UNC | |||
| D32 | 7, M | Tonsil | Follicular | + | + | + | + | R | GCB | I | CT-P | CR, 22 mo | |
| D35 | 6, F | Cervical LN | Diffuse | + | + | − | + | R | GCB | I | CT-P | CR, 63 mo | |
| D46 | 11, M | Tonsil | Follicular and diffuse | + | + | − | + | R | R | GCB | Surgical excision | ||
| D47 | 22, M | Tonsil | Follicular and diffuse | + | + | − | + | R | R | I | CT-A | CR, 24 mo | |
| D48 | 18, F | Tonsil | Follicular and diffuse | − | + | − | + | R | UNC | I | CT-A | CR, 29 mo | |
| D50 | 17, F | Cervical LN | Follicular | − | + | − | + | R | R | Surgical excision | |||
| D51 | 10, F | Tonsil | Follicular and diffuse | + | + | − | + | R | GCB | III | CT-P | CR, 14 mo | |
| D54 | 18, F | Cervical LN | Diffuse | + | + | + | + | R | GCB | III | CT-P | CR, 14 mo | |
| D62 | 17, M | Cervical LN | Follicular | − | + | − | + | R | GCB | II | CT-P | CR, 45 mo | |
| D63 | 14, F | Larynx | Diffuse | + | + | + | + | R | GCB | I | CT-P | CR, 18 mo | |
| D69 | 15, M | Intestine | Follicular and diffuse | − | + | + | + | R | CT-P | CR, 10 mo | |||
| Case | Age (y), gender | Biopsy site | Growth pattern | Immunohistochemistry | FISH | COO NanoString (Lymph2Cx) | Stage* | Treatment | Outcome, follow-up | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD10 | BCL6 | BCL2 | IRF4/MUM1 | IRF4 | IGH | ||||||||
| D1 | 5, F | LN | Follicular and diffuse | + | + | + | + | ||||||
| D2 | 14, M | LN | Follicular | + | + | − | + | R | GCB | Surgical excision | |||
| D15 | 5, F | Tonsil | Diffuse | − | + | + | + | R | R | GCB | II | CT-P | CR, 49 mo |
| D16 | 14, F | Tonsil | Diffuse | − | + | − | + | R | II | CT-P | CR, 99 mo | ||
| D17 | 21, M | Liver | Diffuse | − | + | + | + | R | ABC | I-E | CT-A | CR, 10 mo † | |
| D20 | 17, M | Ileum | Diffuse | + | + | + | + | N | R | UNC | II | CT-P | CR, 72 mo |
| D21 | 8, M | Tonsil | Follicular | + | + | + | + | R | I | CT-P | CR, 36 mo | ||
| D23 | 21, F | Inguinal LN | Diffuse | − | + | + | R | R | GCB | IV-A | CT-A | CR, 40 mo | |
| D31 | 12, F | Cervical LN | Diffuse | − | + | + | + | N | R | UNC | |||
| D32 | 7, M | Tonsil | Follicular | + | + | + | + | R | GCB | I | CT-P | CR, 22 mo | |
| D35 | 6, F | Cervical LN | Diffuse | + | + | − | + | R | GCB | I | CT-P | CR, 63 mo | |
| D46 | 11, M | Tonsil | Follicular and diffuse | + | + | − | + | R | R | GCB | Surgical excision | ||
| D47 | 22, M | Tonsil | Follicular and diffuse | + | + | − | + | R | R | I | CT-A | CR, 24 mo | |
| D48 | 18, F | Tonsil | Follicular and diffuse | − | + | − | + | R | UNC | I | CT-A | CR, 29 mo | |
| D50 | 17, F | Cervical LN | Follicular | − | + | − | + | R | R | Surgical excision | |||
| D51 | 10, F | Tonsil | Follicular and diffuse | + | + | − | + | R | GCB | III | CT-P | CR, 14 mo | |
| D54 | 18, F | Cervical LN | Diffuse | + | + | + | + | R | GCB | III | CT-P | CR, 14 mo | |
| D62 | 17, M | Cervical LN | Follicular | − | + | − | + | R | GCB | II | CT-P | CR, 45 mo | |
| D63 | 14, F | Larynx | Diffuse | + | + | + | + | R | GCB | I | CT-P | CR, 18 mo | |
| D69 | 15, M | Intestine | Follicular and diffuse | − | + | + | + | R | CT-P | CR, 10 mo | |||
CR, complete response; CT-A, chemotherapy with adult schema protocol (R-CHOP/ESHAP); CT-P, chemotherapy with pediatric schema protocol; F, female; LN, lymph node; M, male; N, normal; R, rearrangement; UNC, intermediate/unclassified.
Stage was established according St. Jude/International Pediatric NHL Staging System or Ann Arbor staging system for pediatric and adult patients, respectively.
Patients who had a relapse/progression and needed rescue treatment.
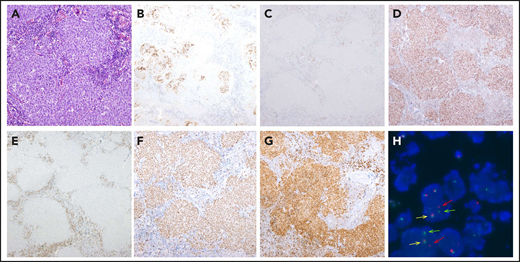
Morphological, immunophenotypic, and genetic features of a LBCL with IRF4 rearrangement (case D62). Architecture effacement by an atypical lymphoid proliferation with nodular growth pattern (A; hematoxylin and eosin) that corresponds to expanded follicles with highly disrupted follicular dendritic cell meshwork (B; CD21). The atypical cells were negative for CD10 (C) and positive for BCL6 (D). BCL2 was positive in the accompanying reactive T cells but negative in the tumor (E), which exhibits a high proliferation rate (F; Ki67). The immunohistochemical study for IRF4/MUM1 (G; MUM1) shows strong and diffuse positivity in the neoplastic proliferation, and FISH with IRF4 break-apart probe shows a signal constellation of 1 colocalization (yellow arrow) and 1 split signal (red and green arrows) consistent with the gene rearrangement (H). Original magnification ×100 (A), ×40 (B-G).
Morphological, immunophenotypic, and genetic features of a LBCL with IRF4 rearrangement (case D62). Architecture effacement by an atypical lymphoid proliferation with nodular growth pattern (A; hematoxylin and eosin) that corresponds to expanded follicles with highly disrupted follicular dendritic cell meshwork (B; CD21). The atypical cells were negative for CD10 (C) and positive for BCL6 (D). BCL2 was positive in the accompanying reactive T cells but negative in the tumor (E), which exhibits a high proliferation rate (F; Ki67). The immunohistochemical study for IRF4/MUM1 (G; MUM1) shows strong and diffuse positivity in the neoplastic proliferation, and FISH with IRF4 break-apart probe shows a signal constellation of 1 colocalization (yellow arrow) and 1 split signal (red and green arrows) consistent with the gene rearrangement (H). Original magnification ×100 (A), ×40 (B-G).
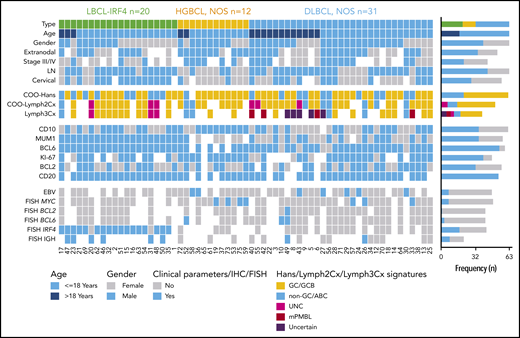
Overview of clinical and histological findings in 63 pediatric and young adult LBCL cases. Each column of the heatmap represents 1 LBCL case and each line a specific analysis. On the right side of the figure, the frequency of the particular result of the analysis is shown. LN, lymph node; UNC, unclassified.
Overview of clinical and histological findings in 63 pediatric and young adult LBCL cases. Each column of the heatmap represents 1 LBCL case and each line a specific analysis. On the right side of the figure, the frequency of the particular result of the analysis is shown. LN, lymph node; UNC, unclassified.
Thirty-one cases (22 males and 9 females; median age, 14 years; range, 1-25 years) were classified as DLBCL, NOS, all with a diffuse pattern of large, mainly centroblastic cells. Most cases showed a GC-phenotype (20/31, 65%) in line with the Lymph2Cx results that showed a GCB profile in 67%, followed by 22% ABC and 11% UNC. MYC breaks were detected in 3 cases, BCL6 rearrangement in 2 cases (supplemental Table 5), and, contrary to adult DLBCL, NOS, only 1 case presented BCL2 translocation. EBV was positive in 5 out of 25 (20%) cases, 4 of which had an ABC phenotype. Seven patients had primary extranodal presentation.
Finally, 12 cases were classified as HGBCL, NOS (8 males and 4 females; median age, 9.5 years; range, 3-23 years), 8 with intermediate features between DLBCL and BL and 4 with blastoid morphology (Figure 3). These cases mainly had a GC phenotype (91%) and were classified as GCB (7/9 cases) by the Lymph2Cx assay. Inmunohistochemically, BCL2 was positive in 5 out of 11 cases (45%) and MYC in 3, but without typical BL morphology (supplemental Figure 4). MYC and BCL6 translocations were detected in 4 cases and 1 case, respectively. No double/triple hits were identified. EBV was detected in 2 cases. Most patients had a primary extranodal presentation (75%) (Figure 2; supplemental Table 6).
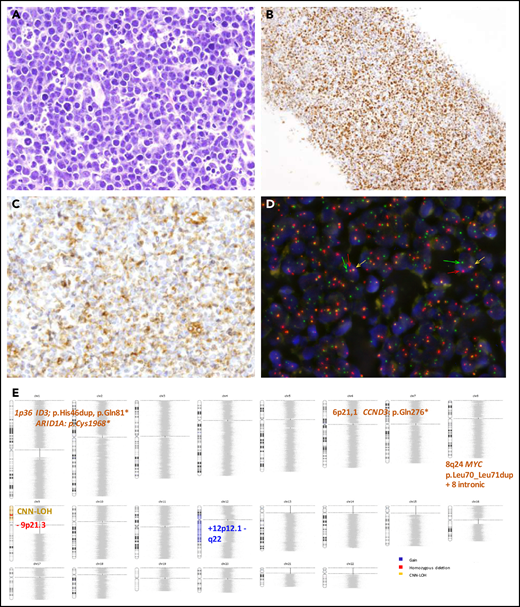
Morphological, immunophenotypic and genetic features of an HGBCL, NOS with MYC rearrangement (Case D59). Hematoxylin and eosin stain (A) depicting mild heterogeneity with certain cellular irregularity of the neoplastic cells that are BCL6 positive (B) with partial expression of BCL2 (C). (D) FISH with MYC break-apart shows a signal constellation of 1 colocalization (yellow arrow) and 1 split signal (green and red arrows). (E) Ideogram of the CN, CNN-LOH, and mutational features of this case. Original magnification ×400 (A,C), ×100 (B).
Morphological, immunophenotypic and genetic features of an HGBCL, NOS with MYC rearrangement (Case D59). Hematoxylin and eosin stain (A) depicting mild heterogeneity with certain cellular irregularity of the neoplastic cells that are BCL6 positive (B) with partial expression of BCL2 (C). (D) FISH with MYC break-apart shows a signal constellation of 1 colocalization (yellow arrow) and 1 split signal (green and red arrows). (E) Ideogram of the CN, CNN-LOH, and mutational features of this case. Original magnification ×400 (A,C), ×100 (B).
A recent gene expression study has recognized PMBL at nonmediastinal sites and/or with atypical clinical presentations.34 To identify any potential PMBL not recognized based on conventional clinicopathological criteria, we investigated the mPMBL signature using the Lymph3Cx assay32 in 39 cases (21 DLBCL, NOS; 14 LBCL-IRF4; and 4 HGBCL, NOS) with available RNA. This analysis predicted 4 DLBCL, NOS as mPMBL. Three of these cases had mediastinal involvement but were not considered initially as PMBL due to concomitant disseminated disease involving bone marrow, lymph nodes, and multiple extranodal sites (supplemental Table 7). However, 1 DLBCL, NOS case predicted as mPMBL had a solitary axillary lymph node without apparent mediastinal involvement. These 4 cases were analyzed as a separate category in subsequent molecular analyses. Finally, the assay predicted 5 DLBCL, NOS, as “uncertain,” ie, with a gene expression signature probability score between DLBCL and PMBL. None of these 5 cases had clinicopathological features of PMBL (supplemental Table 7). Additionally, Lymph2Cx/Lymph3Cx had 100% concordance for COO prediction (Figure 2).
Identification of mutational profiles by targeted NGS
Fifty-five tumors (50 primary, including 22 DLBCL, NOS; 17 LBCL-IRF4; 8 HGBCL, NOS; 3 mPMBL; and 5 relapsed samples) were analyzed by NGS (mean coverage, 447×; range, 28-1439×). After filtering, 496 mutations were identified in 50 out of 55 samples analyzed with a verification rate of 97% (147/151) of the selected variants (supplemental Table 8). A total of 331 variants (67%) were predicted as potential driver mutations (supplemental Methods). After exclusion of mPMBL and relapsed samples, the remaining 47 cases displayed a total of 245 driver mutations with a mean of 5.2 driver mutations per case. The most recurrently mutated genes were IRF4 (14/47, 30%), CARD11 and CCND3 (8/47, 17%), KMT2D, MYC, PIM1, or SOCS1 (6/47, 13%), and FOXO1 (5/47, 11%) (Figure 4A).

Mutational landscape of 47 pediatric and young adult LBCL cases. (A) Bar graphs show mutated genes in >5% of 47 pediatric and young adult primary LBCL cases excluding mPMBL. Each color bar indicates histological subtypes. An asterisk represents mutated genes significantly enriched in 1 of the subtypes. SNV, single-nucleotide variant. (B) A diagram of the relative positions of driver mutations is shown for IRF4, CARD11, CD79B, SOCS1, EZH2, and MYC genes. x-axes indicate amino acid position: IRF4 domains (DBD, DNA-binding domain; IAD, IRF association domain), CARD11 domains (CARD, caspase activation and recruitment domain; SH, Src homology 3 domain), CD79B domains (IG, immunoglobulin-like; ITAM, immunoreceptor tyrosine-based activation motif), SOCS1 domains (ESS, extended SH2 subdomain; KIR, kinase inhibitory region), EZH2 domains (CXC, cysteine-rich domain; SANT, SWI3-ADA2-N-CoR-TFIIIB domains; WDB, WD-40 binding domain), and MYC domains (HLH, helix-loop-helix; LZ, leucine zipper; MBI/II, Myc box I and II; TAD, transactivation domain).
Mutational landscape of 47 pediatric and young adult LBCL cases. (A) Bar graphs show mutated genes in >5% of 47 pediatric and young adult primary LBCL cases excluding mPMBL. Each color bar indicates histological subtypes. An asterisk represents mutated genes significantly enriched in 1 of the subtypes. SNV, single-nucleotide variant. (B) A diagram of the relative positions of driver mutations is shown for IRF4, CARD11, CD79B, SOCS1, EZH2, and MYC genes. x-axes indicate amino acid position: IRF4 domains (DBD, DNA-binding domain; IAD, IRF association domain), CARD11 domains (CARD, caspase activation and recruitment domain; SH, Src homology 3 domain), CD79B domains (IG, immunoglobulin-like; ITAM, immunoreceptor tyrosine-based activation motif), SOCS1 domains (ESS, extended SH2 subdomain; KIR, kinase inhibitory region), EZH2 domains (CXC, cysteine-rich domain; SANT, SWI3-ADA2-N-CoR-TFIIIB domains; WDB, WD-40 binding domain), and MYC domains (HLH, helix-loop-helix; LZ, leucine zipper; MBI/II, Myc box I and II; TAD, transactivation domain).
The number of mutations per case was similar among the 3 subtypes (mean: LBCL-IRF4, 5.2 mutations/case; DLBCL, NOS, 5.8; and HGBCL, NOS, 6.6), but they exhibited different mutational profiles (Figure 4). The most frequently mutated genes in LBCL-IRF4 were IRF4 (76%), CARD11 (35%), and CCND3 (24%). Interestingly, mutations in 3 genes activating the NF-κB pathway (CARD11, CD79B, and MYD88) were observed in 6 out of the 17 analyzed cases. Of note, these 6 cases showed a purely diffuse morphology. The majority of CARD11 mutations (4/6) occurred in the coiled-coil domain, which is known to produce a constitutive NF-κB activation and enhanced NF-κB activity in adult DLBCL.35 All CD79B mutated cases carried a Y197 hot spot mutation affecting the immunoreceptor tyrosine-based activation motif domain (Figure 4B). Mutations on this residue reduce its negative regulation by the kinase LYN.36 MAP2K1 mutations, typically seen in pediatric-type follicular lymphoma, were detected in 2 cases with predominant follicular growth pattern and confirmed IRF4-rearrangement (supplemental Figure 5).37,38 Multiple IRF4 mutations (>4 mutations/case including synonymous variants) were observed in 9 cases, all of which carried the IRF4 rearrangement. These mutations had the pattern of aberrant somatic hypermutation (aSHM) and predominant AID mutational signature (supplemental Results; supplemental Figure 6; supplemental Table 9). In agreement with previous observations,17 8 out of 16 primary LBCL-IRF4 cases investigated carried somatic intronic BCL6 mutations. Of note, these mutations affected the predicted IRF4-binding site in 5 cases (supplemental Table 10).39
Among the 22 DLBCL, NOS cases, the most frequently mutated genes were SOCS1 (27%), KMT2D (23%), and BTG1, EZH2, GNA13, MYD88, and PIM1 (14%), consistent with a predominantly GCB-DLBCL profile but with absence of TNFRSF14 and SGK1 mutations (supplemental Figure 7). Compared with adult DLBCL, NOS, no significant differences were detected in the number of mutations affecting commonly interrogated genes (pediatric/young adult LBCL mean 4.3 vs adult DLBCL 4.8 mutations/case, P < .15). However, we observed a higher frequency of SOCS1 mutations in pediatric and young adult DLBCL, NOS (27% vs 8% respectively, P = .01), and the practical absence of some mutations affecting genes was strongly associated with the definition of established mutational clusters in adult DLBCL, NOS, such as MYD88-L265P, NOTCH1, NOTCH2, BCL2, and SGK1 (supplemental Figure 8A).5,6
Of the 8 HGBCL, NOS cases examined, 4 had mutations in ≥3 genes predominately associated with BL (supplemental Figure 5). The remaining 4 cases had mutations in CARD11 (2 cases) or EZH2 and TNFRSF14 (1 case each) akin to DLBCL, NOS.22,23 Interestingly, MYC mutations clustered around known phosphorylation sites required for the ubiquitination and degradation of MYC protein as previously described (Figure 4B).40 Of note, all MYC mutated cases (5 HGBCL, NOS and 1 DLBCL, NOS) had multiple mutations (>4 mutations/case including intronic and synonymous variants) with an aSHM pattern (supplemental Table 9).40,41 Four out of these 6 cases also carried MYC rearrangement, and in the 2 remaining ones, the presence of cryptic translocations could not be completely ruled out.13,42
We evaluated the presence of recurrent mutated pathways in the different morphological subtypes.26 This analysis showed frequent mutations in chromatin modifiers in HGBCL, NOS, whereas mutations in B-cell differentiation and JAK-STAT pathway genes were more frequently seen in LBCL-IRF4 and DLBCL, respectively (supplemental Figure 9).
The mutational profile of the 3 cases predicted as mPMBL was closer to PMBL than DLBCL, NOS, with mutations in SOCS1, NFKBIE, STAT6, B2M, and CIITA, which appeared to confirm the mPMBL gene expression prediction (supplemental Figure 7).
Finally, the mutational profile of the 3 paired diagnostic-relapse samples analyzed showed marked differences between both biopsy specimens with a total of 13 (mean 4.3, range 0-9) shared and 23 acquired variants (mean 7.7, range 2-16) in the relapsed samples. Additionally, in the 5 relapsed samples available, we identified recurrent KLHL6 and BTG2 mutations (2 cases each) (supplemental Figure 10A).
CN alteration profile
CN analysis detected 302 genetic alterations in 43 out of 49 LBCL primary tumors (mean, 6.2 alterations per case; range, 0-34 alterations) and 45 CNN-LOH in 25 out of 49 cases (supplemental Table 11). Recurrent CN alterations (>15%) included gains of 1q21.2-q42.13, 11q22.3-q25, trisomies 7 and 12, and recurrent losses of 1p36.33-p36.13/TNFRSF14 and 6q21/PRDM1. Frequent CNN-LOH (>10%) affected 17q21.3-q25.3 and 19p13.3-p13.2 regions (supplemental Figure 11). Recurrent homozygous deletions were observed at 19p13.3/CD70 (5 cases) and 9p21.3/CDKN2A (3 cases) in addition to single events in 13q14.2/RB1 and 17q24.1/GNA13 loci. Alteration patterns suggestive of chromothripsis43 were found in 4 out of 49 cases (8%) affecting chromosomes 1, 9, 12, and 13, respectively. Of note, all 4 cases carried MYC, IRF4, or BCL6 translocations.
The 3 LBCL subtypes displayed different CN profiles despite having similar number of aberrations (mean, LBCL-IRF4, 6.2; DLBCL, NOS, 5.8; and HGBCL, NOS, 7.1 alterations per case). LBCL-IRF4 had frequent 17p/TP53 deletions (25%), without gene mutations, and gains of chromosome 7 (45%) and 11q12.3-q25 (35%). DLBCL, NOS had recurrent 2p16 gains targeting REL and 19p13 homozygous deletions targeting CD70 (23% each). HGBCL, NOS had 1q gains (3 cases), similar to BL,30 but also carried trisomies/gains of 7 (4 cases) and 12 (3 cases), and 9p21.3/CDKN2A homozygous deletions (2 cases) (Figure 5).
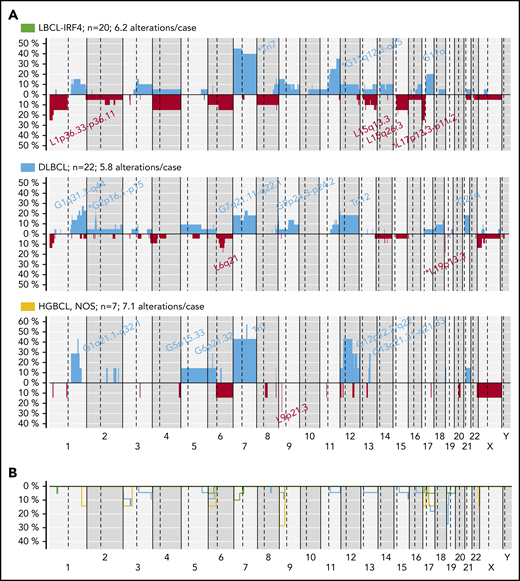
CN profile of pediatric and young adult LBCL cases. (A) Global CN profiles of 20 LBCL-IRF4 cases; 22 DLBCL, NOS cases; and 7 HGBCL, NOS cases. x-axis indicates chromosomes from 1 to Y and p to q. The vertical axis indicates frequency of each genomic aberration among the analyzed cases. Gains are depicted in blue and losses in red. Most frequently recurrent regions are indicated for LBCL-IRF4; DLBCL, NOS (>20%); and HGBCL, NOS (≥2 cases). Asterisks indicate significant differences between LBCL-IRF4 and DLBCL (Fisher’s test, P < .05). (B) Comparative plot of CNN-LOH among the 3 morphological groups described above. Green identifies LBCL-IRF4; blue DLBCL, NOS; and yellow HGBCL, NOS.
CN profile of pediatric and young adult LBCL cases. (A) Global CN profiles of 20 LBCL-IRF4 cases; 22 DLBCL, NOS cases; and 7 HGBCL, NOS cases. x-axis indicates chromosomes from 1 to Y and p to q. The vertical axis indicates frequency of each genomic aberration among the analyzed cases. Gains are depicted in blue and losses in red. Most frequently recurrent regions are indicated for LBCL-IRF4; DLBCL, NOS (>20%); and HGBCL, NOS (≥2 cases). Asterisks indicate significant differences between LBCL-IRF4 and DLBCL (Fisher’s test, P < .05). (B) Comparative plot of CNN-LOH among the 3 morphological groups described above. Green identifies LBCL-IRF4; blue DLBCL, NOS; and yellow HGBCL, NOS.
Compared with adult DLBCL, NOS, pediatric and young adult DLBCL, NOS had a similar CN profile without any specific alteration but significantly lower levels of genetic complexity (mean, 5.8 vs 20 CN alterations; P < .01) (supplemental Figure 8B). Our current pediatric and young adult series lacked alterations present in adults such as 6q13-q14.1/TMEM30A and 6q22.1-q25.3/TNFAIP3 deletions as well as those typically associated with ABC-DLBCL as 9p21.3/CDKN2A and 17p13.3-p11.2/TP53 losses, which probably reflects the predominance of GCB cases in our cohort. In fact, these differences were not observed when compared only to adult BCL2-negative GCB-DLBCL. Nevertheless, results should be taken with caution, since different CN platforms were used.
Finally, analysis of 3 paired diagnostic-relapsed biopsy specimens showed the acquisition of a mean of 15 additional events (range, 12-16) in the relapsed samples. Recurrent alterations in these cases included gains of 1q21.2-q41 (MDM4), 12p13.3-q21.1, and 18q22.2-q23 and biallelic inactivation of 19p13/CD70, which was also present in both samples (diagnosis/relapse) in the 3 cases (supplemental Figure 10B) and even in a second relapse in 1 case.
Gene expression patterns
Differential gene expression analysis between LBCL-IRF4 (n = 11) and DLBCL, NOS (n = 10) identified 48 differentially expressed genes (log2 fold change greater than ±1 and false discovery rate <.05), including 14 NF-κB target genes (29%; eg, IRF4, LTF, and CSF1), which suggests a deregulation of this pathway in LBCL-IRF4 (http://www.bu.edu/nf-kb/gene-resources/target-genes/) (supplemental Figure 12; supplemental Table 12). No gene expression differences were observed between LBCL-IRF4 cases with and without CD79B or CARD11 mutations.
Prognostic value of clinical and molecular features
The 5-year EFS of the 46 LBCL patients with available follow-up was 68.4%. All received chemotherapy as first-line treatment, including rituximab in 35% of the patients with no differences in EFS (log-rank test P = .75). Complete response was achieved in 83% of patients, whereas 8 died of disease. Cases predicted as ABC-DLBCL had significantly worse 5-year EFS compared with GCB-DLBCL patients (26% vs 74% P = .002) (Figure 6; supplemental Figure 13), even when the LBCL-IRF4 cases were excluded from the comparison (30% vs 68%, P = .033). Interestingly, the clinical outcome of GCB-DLBCL was similar to LBCL-IRF4, whereas ABC-DLBCL and HGBCL, NOS had significantly worse 5-year EFS (78% vs 48%; P = .005). Similarly, high lactate dehydrogenase (LDH) levels; a high number of CN alterations (>10), including cases with chromothripsis-like patterns; TP53 and MYC mutations; gains of 2p16/REL and 1q21-q44/MDM4; and biallelic inactivation of 19p13/CD70 correlated with a significant lower EFS rate (Figure 6; supplemental Figure 13).
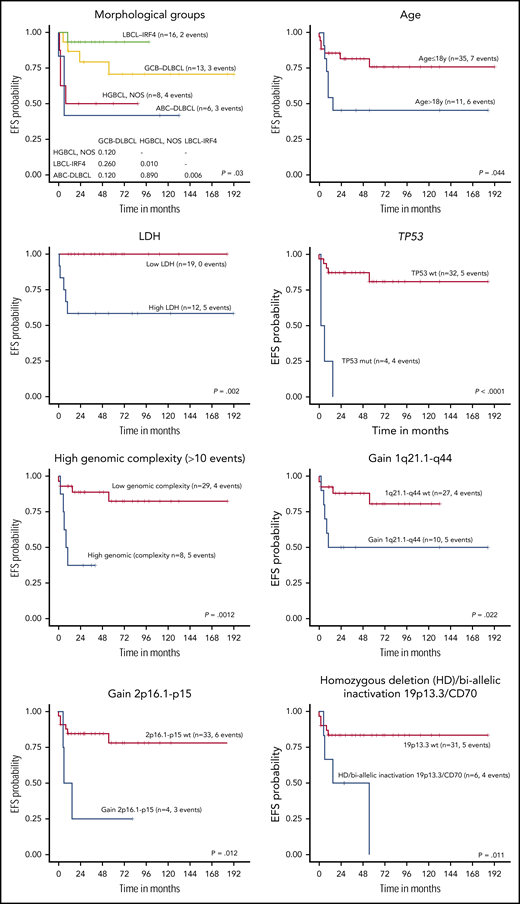
EFS of 45 pediatric and young adult LBCL cases according to morphological and molecular subtypes, age, LDH levels, and specific molecular features. wt, wild type.
EFS of 45 pediatric and young adult LBCL cases according to morphological and molecular subtypes, age, LDH levels, and specific molecular features. wt, wild type.
When patients were examined according to age, pediatric patients (≤18 years) had better 5-year EFS than young adults (19-25 years) (76% vs 46%, log-rank test P = .044) (Figure 6; supplemental Figure 13). Young adults had more frequent BCL6 rearrangements, high genetic complexity, and chromothripsis-like patterns (supplemental Table 13). Finally, when the clinical parameters were compared among the 3 different lymphoma entities, HGBCL, NOS presented more frequently in primary extranodal sites and had a higher incidence of MYC rearrangements (supplemental Table 14).
Discussion
Genomic studies of adult LBCL have revealed a large number of somatic mutations and structural alterations related to the pathogenesis of these diseases.3,4,7 However, the genetic landscape of these tumors in pediatric populations is poorly known. In this study, through an integrative targeted NGS, CN, and transcriptome data analyses, we show that pediatric and young adult LBCLs are a heterogeneous group of tumors including different entities with specific molecular profiles and clinical behavior. A better understanding of these differences is relevant to design management strategies more adapted to the particular biological behavior of these tumors.
LBCL-IRF4 was recently recognized as a specific entity genetically characterized b IRF4 translocation, clinical presentation localized in the head and neck or abdominal regions, and a favorable outcome after chemotherapy.17,19 We have now expanded these observations to show that these tumors have a distinct molecular profile characterized by frequent mutations in IRF4 and NF-κB-related genes (CARD11, CD79B, and MYD88) and overexpression of downstream target genes of the NF-κB pathway. These findings are intriguing, because most of these tumors have a GC phenotype and gene expression profile, whereas mutations in these genes and NF-κB activation have mainly been found in ABC-DLBCL in adults.5 The activation of the NF-κB pathway in these tumors may be also related to the IRF4 overexpression.44 The presence of multiple mutations affecting the IRF4 gene with an aSHM pattern appears to be a hallmark of the IRF4 translocation. Further studies are needed to define the potential functional effect of these mutations. Morphologically, LBCL-IRF4 may have predominantly diffuse or follicular pattern. Interestingly, CARD11 mutations were seen exclusively in cases with diffuse growth pattern, whereas MAP2K1 mutations, characteristic of pediatric-type follicular lymphoma,37,38 were detected in 2 cases with a predominantly follicular pattern (supplemental Figure 8), suggesting that the underlying mutational profile may influence the morphological features of the tumors. No differences in terms of CN mutational profile, including IRF4 mutations and IRF4 expression at the RNA and protein level (supplemental Figure 3), were seen in the 2 LBCL-IRF4 cases with an IGH split without concomitant IRF4 breaks, confirming the idea that these tumors belong to the same entity. The presence of cryptic IRF4 translocations cannot be excluded.45 The outcome of LBCL-IRF4 was very favorable, but most of the patients were treated with aggressive pediatric or adult-type chemotherapy protocols. The identification of the translocation and the related mutational profile may be relevant to identify these patients and design management strategies better suited to the biology of the tumors.17,19
The genetic and expression profile of our DLBCL, NOS was relatively similar to that previously observed in pediatric cohorts with a predominance of GCB-DLBCL, low CN complexity, few MYC and BCL6 rearrangements, and, in contrast to adults, the virtual absence of BCL2 translocations (supplemental Table 15).46-48 Our study expands these observations, showing that these cases have a mutational profile similar to adult DLBCL with predominance of mutations in GCB-DLBCL–related genes (supplemental Figures 5 and 8). Nevertheless, pediatric and young adult DLBCL had higher frequency of SOCS1 mutations and virtually lacked MYD88-L265P, NOTCH1, NOTCH2, BCL2, and SGK1 mutations that have been associated with the definition of established mutational clusters in adult DLBCL.5,6 Further studies using whole-exome/genome approaches may expand the genomic profile of alterations of these tumors.
The application of the Lymph3Cx in our DLBCL, NOS cohort recognized 4 cases predicted as mPMBL with an atypical clinical presentation for this entity.32 Although 3 of these patients had mediastinal lymph node involvement, they also had disseminated disease including bone marrow and extranodal involvement. Intriguingly, 1 case predicted as mPMBL only had axillary nodal involvement. The mutational profile of these cases was consistent with PMBL, including NFKBIE mutations recently associated with poor outcome in these tumors.49 These observations, together with similar cases recently described in adults, suggest that a subset of DLBCL, NOS in pediatric and young adult populations may correspond to PMBL.34,50
The genetic features of HGBCL, NOS were heterogeneous. Four out of the 8 molecularly investigated cases had mutational profile closer to BL (supplemental Figure 5), with concomitant MYC rearrangements and 1q21-q31 gains identified in 2 cases each (Figure 3E; supplemental Figure 4). Nevertheless, those cases did not have the typical BL morphology and expressed strong BCL2 or MUM1. Similar to HGBCL, NOS in adults,22,23 no TCF3 mutations were seen in our cases. Other HGBCL, NOS cases had mutational profiles closer to GCB-DLBCL with TNFRSF14, CARD11, and EZH2 mutations and lacked MYC translocations. Of note, genes frequently mutated in BLL-11q, such as BTG2, ETS1, and EP300,15,16 were significantly absent in both DLBCL and HGBCL, NOS (P < .05), suggesting that they correspond to different entities (supplemental Figure 5).
Regarding prognostic aspects, advanced stage, high LDH, and combined bone marrow and central nervous system disease have been significantly associated with unfavorable outcome in pediatric mature B-cell lymphomas, whereas the adverse prognosis of 8q24-MYC rearrangements and ABC-COO is still controversial.24,27 In our series, we found the prognostic value of several clinical and molecular features such as age >18 years, high LDH levels, and ABC-subtype, as seen in adult populations. We also found that TP53 mutations, high genetic complexity, including chromothripsis, and gains in 1q21-q44/MDM4 conferred poor EFS.
In conclusion, LBCLs in the pediatric and young adult population are a heterogeneous group of tumors with distinct genomic and mutational alterations. LBCL-IRF4 reveals a GC phenotype with frequent mutations in IRF4, NF-κB-related genes (CARD11, CD79B, and MYD88), and overexpression of genes of the NF-κB pathway, whereas DLBCL, NOS cases in this population are predominantly of GCB subtype. Our study also suggests that PMBL may present with disseminated disease, and ancillary tools may recognize these cases, with implications for treatment. The integration of molecular and genetic studies may improve the classification of LBCL in pediatric and young adult populations and provide parameters for risk stratification.
The CN and gene expression data reported in this article have been deposited to the GEO database under accession number GSE128294. Sequencing data have been deposited to the European Nucleotide Archive under accession number ERP114095.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the centers of the Sociedad Española de Hematología y Oncología Pediátricas that submitted cases for consultation, as well as Noelia Garcia, Silvia Martín, and Helena Suarez for their excellent technical assistance. They also thank Miguel Angel Piris from Fundación Jimenez Díaz for performing immunohistochemical analyses and Pedro Jares and Magda Pinyol from Hospital Clínic, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) for their support with gene expression analyses. The authors are indebted to the IDIBAPS Genomics Core Facility and the HCB-IDIBAPS Biobank-Tumor Bank and Biobanc de l’HospitaI Infantil Sant Joan de Déu, Hospital Universitario Virgen del Rocío-Instituto de Biomedicina de Sevilla Biobank (ISCIII-Red de Biobancos PT13/0010/0056), all 3 integrated in the National Network Biobanks of ISCIII, for the sample and data procurement.
This work was supported by Asociación Española Contra el Cáncer (AECC CICPFI6025SALA), Fondo de Investigaciones Sanitarias Instituto de Salud Carlos III (Miguel Servet Program CP13/00159 and PI15/00580 to I.S.), Spanish Ministerio de Economía y Competitividad (SAF2015-64885-R to E.C.), Generalitat de Catalunya Suport Grups de Recerca (2017-SGR-1107 to I.S. and 2017-SGR-1142 to E.C.), SPECS II grant (National Cancer Institute 1U01CA157581), and the European Regional Development Fund (Una manera de fer Europa). J.E.R.-Z. was supported by a fellowship from Generalitat de Catalunya AGAUR FI-DGR 2017 (2017 FI_B01004). E.C. is an Academia Researcher of the Institució Catalana de Recerca i Estudis Avançats of the Generalitat de Catalunya. This work was developed at the Centro Esther Koplowitz, Barcelona, Spain. The group is supported by Acció instrumental d’incorporació de científics i tecnòlegs PERIS 2016 (SLT002/16/00336 to Noelia Garcia) from Generalitat de Catalunya.
Authorship
Contribution: J.E.R.-Z. performed research, analyzed data, and wrote the manuscript; B.G.-F. performed morphological diagnosis, analyzed data, and wrote the paper; F.N., J.S.-V., I.M.-G., G.C., A.E., A. Maguire, and C.R. performed research and analyzed data; O.B., M.G.-P., A.G., M.S., D.A., C.B., F.G.-B., G.T., A. Mozos, L.M.R., L.Q.-M., and E.S.J. reviewed and interpreted pathological data; V.C., M. Andrés, M. Andión, I.A., M.S.d.I., C.S., S.G., J.V.-A., R.F.-D., A.R.-D., V.P., M.T., P.S.-P., I.D., A.L.-G., P.G., and M.J.O. reviewed and interpreted clinical data; E.C. performed morphological analysis, designed research, and wrote the manuscript; and I.S. performed research, analyzed data, designed research, and wrote the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: E.C. and L.M.R. are co-inventors of the Lymph2Cx and Lymph3Cx gene expression profiling assay used in this study. The remaining authors declare no competing financial interests.
Correspondence: Itziar Salaverria, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Rosselló 149-153, 08036 Barcelona, Spain; e-mail: isalaver@clinic.cat.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal