

TO THE EDITOR:
Targeted therapies have substantially altered the natural history of chronic lymphocytic leukemia (CLL). Venetoclax, an inhibitor of BCL2 apoptosis regulator (BCL2), shows considerable efficacy, including in patients who have relapsed after ibrutinib.1 In the ibrutinib-refractory setting, overall response rate to venetoclax is 65%, with median progression-free survival of 23.5 months.1 This progression-free survival is considerably lower than in patients not previously treated with ibrutinib,2 suggesting potential distinct resistance mechanisms in this population.
Resistance to venetoclax has been associated with G101V mutations in BCL2 in up to 50% of cases.3,4 Other BCL2 mutations, including D103Y, have also been identified in combination with G101V. We investigated ibrutinib-refractory, venetoclax-resistant patients to determine whether BCL2 mutations were common in this population.
Blood samples from CLL patients at The Ohio State University Comprehensive Cancer Center (OSUCCC) were collected under institutional review board–approved protocols. Sample preparation and sequencing methods are in supplemental Methods (available on the Blood Web site).
We identified 24 patients, consented to a tissue-banking protocol at the OSUCCC, who had been treated with ibrutinib, relapsed, and then were subsequently treated with single-agent venetoclax given continuously until progression. Thirteen of these patients developed disease progression, and 11 patients were included in this analysis because of the availability of baseline and relapse blood samples. Clinical characteristics are in Table 1. All patients had high-risk disease, including 10 with unmutated IGHV, 10 with complex karyotype, and 7 with TP53 alterations. Median PFS was 22 months (range, 2-33 months). Richter transformation occurred in 3 patients; 8 patients had progressive CLL. Following progression on venetoclax, patients were primarily treated on clinical trials with novel agents. Two patients with Richter transformation survived 2 months following transformation, and 1 survived for 17 months. Among the 8 patients with CLL progression, 4 were alive at last follow-up (24-42 months postvenetoclax), 1 had withdrawn from follow-up, and 3 had died 6, 10, and 24 months postvenetoclax.
Clinical characteristics and outcomes of CLL patients with venetoclax resistance after BTK-inhibitor treatment
| Pt no. | Sex | No. of prior therapies | IgVH status | Complex karyotype* | MYC | Tris12 | del13q | ATM | p53 | Age at venetoclax initiation, y | Months on venetoclax | Progression type | Survival after progression, mo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 4 | Unmutated | Yes | Yes | Yes | Yes | 74 | 25 | CLL type | NA | ||
| 2 | M | 13 | Unmutated | No | Yes | 53 | 22 | CLL type | NA | ||||
| 3 | M | 2 | Unmutated | Yes | Yes | Yes | 66 | 24 | CLL type | NA | |||
| 4 | M | 7 | Unmutated | Yes | Yes | Yes | 69 | 24 | CLL type | NA | |||
| 5 | M | 5 | Unmutated | Yes | 57 | 13 | CLL type | 6 | |||||
| 6 | M | 6 | Unmutated | Yes | Yes | 79 | 22 | CLL type | 10 | ||||
| 7† | M | 5 | Unmutated | Yes | Yes | Yes | Yes | 59 | 2 | CLL type | NA | ||
| 8 | M | 8 | Unmutated | Yes | Yes | 72 | 33 | CLL type | NA | ||||
| 9 | F | 6 | Unmutated | Yes | Yes | Yes | 59 | 19 | Richter | 2 | |||
| 10 | M | 6 | Unmutated | Yes | Yes | 55 | 6 | Richter | 2 | ||||
| 11 | F | 8 | Mutated | Yes | Yes | Yes | Yes | Yes | Yes | 65 | 4 | Richter | 17 |
| Pt no. | Sex | No. of prior therapies | IgVH status | Complex karyotype* | MYC | Tris12 | del13q | ATM | p53 | Age at venetoclax initiation, y | Months on venetoclax | Progression type | Survival after progression, mo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 4 | Unmutated | Yes | Yes | Yes | Yes | 74 | 25 | CLL type | NA | ||
| 2 | M | 13 | Unmutated | No | Yes | 53 | 22 | CLL type | NA | ||||
| 3 | M | 2 | Unmutated | Yes | Yes | Yes | 66 | 24 | CLL type | NA | |||
| 4 | M | 7 | Unmutated | Yes | Yes | Yes | 69 | 24 | CLL type | NA | |||
| 5 | M | 5 | Unmutated | Yes | 57 | 13 | CLL type | 6 | |||||
| 6 | M | 6 | Unmutated | Yes | Yes | 79 | 22 | CLL type | 10 | ||||
| 7† | M | 5 | Unmutated | Yes | Yes | Yes | Yes | 59 | 2 | CLL type | NA | ||
| 8 | M | 8 | Unmutated | Yes | Yes | 72 | 33 | CLL type | NA | ||||
| 9 | F | 6 | Unmutated | Yes | Yes | Yes | 59 | 19 | Richter | 2 | |||
| 10 | M | 6 | Unmutated | Yes | Yes | 55 | 6 | Richter | 2 | ||||
| 11 | F | 8 | Mutated | Yes | Yes | Yes | Yes | Yes | Yes | 65 | 4 | Richter | 17 |
ATM, ataxia telangectasia mutation; F, female; IgVH, mutation status of the immunoglobulin heavy chain variable region; M, male; NA, not applicable; Pt no., patient number.
Defined as ≥3 abnormalities on CpG oligonucleotide-stimulated karyotype.
Patient also received acalabrutinib prior to venetoclax.
As BCL2 mutations are described to occur at low allele frequencies,3 we investigated BCL2, Bruton tyrosine kinase (BTK), and phospholipase C γ2 (PLCG2) before venetoclax and at clinical progression in all patients using an ultradeep approach (Figure 1; supplemental Table 1). We identified multiple known and novel BCL2 mutations at CLL progression. The known G101V (variant allele frequency [VAF], 9.4%) and a novel A113G (VAF, 31.7%) variant were seen at resistance in 1 patient (patient 4). Mutations in other patients included F104L (VAF, 29.2%) in patient 3, and a novel in-frame insertion of 12 nucleotides resulting in p.Arg107_Arg110dup in 2 patients with CLL-type progression (VAFs, 0.4% and 5%), and 1 patient with Richter transformation (VAF, 0.4%). This internal tandem duplication is in a short repetitive sequence and may be incorrectly rejected by variant callers. Additional mutations occurring at low VAFs were L119V (VAF, 0.2%) and F104L (VAF, 0.3%), both detected in the same patient with the A113G and G101V variants, and p.F104S (0.4%) and p.G101A (1%) coexisting with the p.Arg107_Arg110dup in patient 6. Altogether, targeted deep sequencing of BCL2 detected largely nonrecurring and previously unreported BCL2 mutations clustered around codon 101 in 5 of 11 patients. These were predominantly not present at venetoclax initiation. However, in a single patient with Richter transformation progression, the rare5 allele A61T was present at 50% prior to venetoclax and decreased to 27% at relapse, suggesting a possible copy-number alteration at this locus.
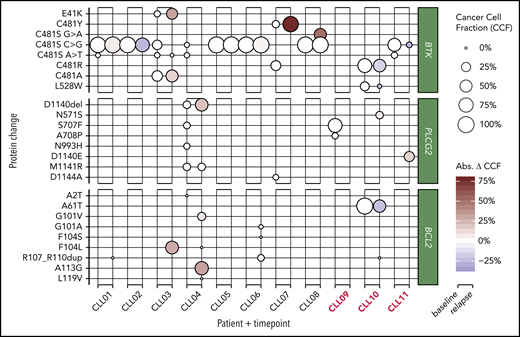
Bubble plots of prevenetoclax and postvenetoclax CCFs of mutations in BTK, PLCG2, and BCL2. Bubble size represents cancer cell fraction (CCF; 0% to 100%), and relapse bubble color encodes absolute gain or loss of clone size. Three patients with Richter-type progression are highlighted in red. CCF was adjusted for patient X-chromosome ploidy. Abs. Δ CCF, absolute change in cancer cell fraction.
Bubble plots of prevenetoclax and postvenetoclax CCFs of mutations in BTK, PLCG2, and BCL2. Bubble size represents cancer cell fraction (CCF; 0% to 100%), and relapse bubble color encodes absolute gain or loss of clone size. Three patients with Richter-type progression are highlighted in red. CCF was adjusted for patient X-chromosome ploidy. Abs. Δ CCF, absolute change in cancer cell fraction.
Using the same approach, we detected BTK and/or PLCG2 mutations in all patients prior to venetoclax (Figure 1; supplemental Table 1). Among the 8 patients with CLL-type progression on venetoclax, 5 (63%) had C481S mutations prevenetoclax and postvenetoclax at nearly identical VAFs. One also acquired an alanine variant in addition to an existing C481S at the same locus. One patient without C481S had significant outgrowth of C481Y (13% to 91% VAF) in the context of C481R loss. In addition, we detected several novel BTK and PLCG2 mutations that persisted or increased throughout venetoclax treatment. Patient 3, with 2 C481S clones (c.1441T>G, c.1442G>C) as well as C481A due to the trinucleotide multiple nucleotide polymorphism c.1441_1443TGC>GCT, showed gain in the multiple nucleotide polymorphism and loss of the smaller C481S clone after venetoclax. This patient additionally showed clonal gain of BTK E41K mutation from 8% to 40% over the course of venetoclax treatment. This mutation is known to constitutively activate BTK, but has not been previously reported in patients. In patient 4, a PLCG2 clone bearing a novel in-frame deletion D1140del showed an increase from 7% to 28% during therapy. Patient 7 uniquely appeared to experience the emergence of a dominant C481S clone. Altogether, these patterns suggest a variety of complex B-cell receptor (BCR) pathway–related mechanisms in patients with CLL-type progression. Indeed, some tumors may have generated multiple signal-enhancing mutations in the BCR pathway as a mechanism of progression during venetoclax treatment.
In contrast, in patients with Richter transformation, BTK and PLCG2 clones generally decreased throughout venetoclax treatment, including a novel NP_000052:p.L528W detected at 23% prior to venetoclax and absent at time of progression. Exceptions were PLCG2 N571S and D1140E, with increased frequencies at relapse compared with baseline, suggesting distinct and variable escape mechanisms.
Although targeted therapies have substantially expanded treatment options for CLL, challenges remain; development of resistance to a targeted agent is one of the most critical at this time. Recent data suggest that similar to ibrutinib, resistance to venetoclax can be mediated by the acquisition of point mutations in the target gene.3,4 However, none of the patients examined in the seminal paper had been previously treated with a BTK inhibitor, in contrast to how venetoclax is often used in clinical practice. Our data from a small population suggest that this mechanism of resistance is less common in patients with prior BTK inhibitor exposure.
Using targeted deep sequencing, we detected multiple additional new somatic mutations clustering around codon 101, including in a patient with Richter transformation. The murine ortholog of F104L has been observed in a lymphoma model,6 but to our knowledge has never been reported in venetoclax-treated patients. We also found a novel in-frame internal tandem duplication present in 3 separate patients. Contrasting the patterns in patients with CLL-type progression, an A61T variant was already present before venetoclax and decreased in frequency during 6 months of treatment in a patient with Richter transformation. Our data altogether suggest different and unique patterns of resistance in patients with CLL-type progression vs those with Richter transformation, similar to what has been observed during ibrutinib treatment.7,8 Interestingly, previous sequencing in a cohort of high-risk CLL patients comparing peripheral blood to lymph node samples did not detect any mutations in BCL family members.9 This cohort, however, was enriched for patients with Richter transformation, and we found only 1 treatment-emergent BCL2 mutation in 1 of 3 Richter patients using an approach with higher coverage.
To our knowledge, this is the first study to describe resistance mechanisms to BCL2 inhibition in the context of ibrutinib-resistance mutations. Conspicuously unlike other reports of new BCL2 mutations in which the classical G101A dominates,10-12 we observed G101A/V in just 2 patients. Our data emphasize that an analysis of clonal dynamics associated with emerging gold-standard treatments for CLL is essential for clinical management of these patients, and that the sequence of these therapies may influence patterns of resistance. Notably, we observe a rapid proliferation of BCR-pathway mutations in several patients during venetoclax therapy in the absence of BTK inhibition, providing rationale for combination therapies. Further studies are needed to determine interactions and dynamics among BTK, PLCG2, and BCL2 clones, especially in the context of alternative cell-intrinsic and microenvironmental-mediated resistance mechanisms.
The sequencing data reported in this article have been deposited in the National Center for Biotechnology Information Sequence Read Archive (BioProject PRJNA577132).
The online version of this article contains a data supplement.
Acknowledgments
The authors are grateful to the patients who provided blood for the above studies and to The Ohio State University Comprehensive Cancer Center (OSUCCC) Leukemia Tissue Bank for sample procurement.
This work was supported by National Institutes of Health (NIH) National Cancer Institute (NCI) grants 5P30CA016058-43, 5R01CA197870 (J.A.W.), and 5R35CA197734 (J.C.B.), and by a grant of equipment from StorageReview (J.S.B.). The OSUCCC Leukemia Tissue Bank was supported by NIH NCI grant P30 CA016058.
K.A.R. is a Scholar in Clinical Research of the Leukemia & Lymphoma Society.
Authorship
Contribution: F.L. performed research, analyzed data, and wrote the manuscript; K.L. performed research, analyzed data, and wrote portions of the manuscript; C.T.G., H.G.O., D.S., S.T., and S.A.Y. analyzed data; S.O. performed research and analyzed data; T.-J.D., A.L., G.L., S.M., and A.N. performed research; K.A.R. and J.C.B. accrued patients and contributed data; J.A.W. and J.S.B. designed and supervised the study, accrued patients, analyzed data, and wrote the manuscript; and all authors edited and approved the final version of the manuscript.
Conflict-of-interest disclosure: J.A.W. receives research funding from AbbVie, Janssen, Loxo, Karyopharm, and Morphosys, and has performed consulting for Janssen and Pharmacyclics. J.S.B. has received consulting fees from AbbVie, AstraZeneca, and KITE Pharma. K.A.R. receives research funding from Genentech, AbbVie, and Janssen and has consulted for Acerta Pharma. The remaining authors declare no competing financial interests.
Correspondence: Jennifer A. Woyach, The Ohio State University, 455D Wiseman Hall, 410 W. 12th Ave, Columbus, OH 43210; e-mail: jennifer.woyach@osumc.edu.
