

Key Points
CDK6 silencing suppresses Ph+ ALL more effectively than CDK4/6 inhibition, supporting the role of kinase-dependent and independent mechanisms.
CDK6-selective PROTACs suppress Ph+ ALL ex vivo and in mice.

Abstract
Expression of the cell cycle regulatory gene CDK6 is required for Philadelphia-positive (Ph+) acute lymphoblastic leukemia (ALL) cell growth, whereas expression of the closely related CDK4 protein is dispensable. Moreover, CDK6 silencing is more effective than treatment with the dual CDK4/6 inhibitor palbociclib in suppressing Ph+ ALL in mice, suggesting that the growth-promoting effects of CDK6 are, in part, kinase-independent in Ph+ ALL. Accordingly, we developed CDK4/6–targeted proteolysis-targeting chimeras (PROTACs) that inhibit CDK6 enzymatic activity in vitro, promote the rapid and preferential degradation of CDK6 over CDK4 in Ph+ ALL cells, and markedly suppress S-phase cells concomitant with inhibition of CDK6-regulated phospho-RB and FOXM1 expression. No such effects were observed in CD34+ normal hematopoietic progenitors, although CDK6 was efficiently degraded. Treatment with the CDK6-degrading PROTAC YX-2-107 markedly suppressed leukemia burden in mice injected with de novo or tyrosine kinase inhibitor–resistant primary Ph+ ALL cells, and this effect was comparable or superior to that of the CDK4/6 enzymatic inhibitor palbociclib. These studies provide “proof of principle” that targeting CDK6 with PROTACs that inhibit its enzymatic activity and promote its degradation represents an effective strategy to exploit the “CDK6 dependence” of Ph+ ALL and, perhaps, of other hematologic malignancies. Moreover, they suggest that treatment of Ph+ ALL with CDK6-selective PROTACs would spare a high proportion of normal hematopoietic progenitors, preventing the neutropenia induced by treatment with dual CDK4/6 inhibitors.
Introduction
Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL) is a poor-prognosis malignancy driven by the BCR-ABL1 isoform p190 or, less frequently, p210, both of which have constitutive tyrosine kinase activity.1,2 Tyrosine kinase inhibitors (TKIs) are now the mainstay of Ph+ ALL therapy. In combination with standard chemotherapy, TKIs have markedly improved the outcome of patients with Ph+ ALL.3,4 However, resistance to TKIs develops frequently, causing leukemia relapse that results in a <5-year overall survival in ∼50% of patients.5 Thus, new therapies are required to address relapsed/TKI-resistant Ph+ ALL.
We have shown that Ph+ ALL cells are dependent on CDK6 expression, whereas CDK4 is dispensable due, in part, to its exclusively cytoplasmic localization.6 Inhibition of CDK6 activity with the CDK4/6 inhibitor palbociclib7 suppressed growth of Ph+ ALL cells ex vivo6,8 and in mice.6 However, the effect was primarily cytostatic, with leukemia growth resuming rapidly upon palbociclib discontinuation.
In addition to regulating the G1-S transition, CDK6 exhibits growth-promoting functions in hematologic malignancies. Several of these functions seem to be kinase-independent and are due to the interaction of CDK6 with transcriptional regulators enhancing or inhibiting gene expression.9-15 In T-cell ALL, CDK6 exerts kinase-dependent prosurvival effects by redirecting the glycolytic pathways to the increased production of the antioxidants reduced NADPH and glutathione and decreased levels of reactive oxygen species.16 Thus, agents that reduce CDK6 expression should suppress Ph+ ALL through kinase-dependent and independent mechanisms and, compared with dual CDK4/6 inhibitors, spare normal hematopoietic progenitors that rely on CDK4 and CDK6 for their growth.
Designing CDK6-selective adenosine triphosphate (ATP) competitive inhibitors devoid of activity toward CDK4 is challenging because the ATP-binding domain of human CDK4 and CDK6 is virtually identical, with the exception of Glu21 of CDK6 that is replaced by Val14 in CDK4. Moreover, such compounds would not inhibit the kinase-independent effects of CDK6.
Unlike the ATP-binding pocket, the solvent-exposed surface of CDK4/6 is less conserved between the 2 proteins, perhaps explaining why these 2 kinases have preferred binding partners.17,18 We therefore undertook a discovery effort to identify proteolysis-targeting chimeras (PROTACs) that would potently inhibit CDK6 kinase activity and selectively degrade CDK6 over CDK4.
PROTACs are molecules comprising a ligand for the protein of interest and an E3 ligase–recruiting ligand that are connected by a linker, allowing recruitment of E3 ligases for optimal ubiquitination and proteasomal degradation.19,20 Compared with traditional competitive inhibitors that are required to bind continuously to a target protein to inhibit its activity, a PROTAC requires less drug exposure due to its catalytic mechanism, where it binds a substrate, recruits an E3 ligase for ubiquitination/proteasomal degradation, and is released to repeat the cycle.21 Moreover, a CDK6-degrading PROTAC would prevent the compensatory increase in CDK6 expression seen with clinically used CDK4/6 enzymatic inhibitors.22,23
We report here the development of CDK4/6–targeted PROTACs that degrade rapidly and preferentially CDK6 over CDK4, inhibit proliferation of Ph+ ALL cells, and suppress patient-derived Ph+ ALL in mice.
Methods
A complete Methods section is provided in the supplemental Material (available on the Blood Web site).
Cell lines, Ph+ primary ALL samples, and cell cultures
The SUP-B15 (Ph+ ALL) and the SEM (MLL-rearranged [MLL-AF4] ALL) cell lines were purchased from ATCC (Manassas, VA); the BV173 cell line (Ph+ chronic myeloid leukemia–lymphoid blast crisis24 was provided by N. Donato (National Institutes of Health, Bethesda, MD); the Ph-like ALL MUTZ-5 and MHH-CALL-4 cell lines were kindly provided by M. Carroll (University of Pennsylvania, Philadelphia, PA); and the 697 t(1;19) ALL cell line was kindly provided by C. Croce (The Ohio State University Comprehensive Cancer Center, Columbus, OH). Cell lines were cultured in Iscove modified Dulbecco medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin-streptomycin, and 2 mmol/L l-glutamine at 37°C, 5% carbon dioxide, and tested for mycoplasma every 3 months as previously described.6
Primary adult human Ph+ ALL cells were provided by Luke F. Peterson (University of Michigan, Ann Arbor, MI), Alessandro Rambaldi (Università Statale Milano, Milan, Italy), and by the Division of Hematological Malignancies of Thomas Jefferson University (Philadelphia, PA). The TKI-resistant Ph+ ALL sample (#557) has the T315I ABL1 kinase domain mutation.25 Primary Ph+ ALL cells were cultured in StemSpan SFEM (Stemcell Technologies, Vancouver, BC, Canada) supplemented with stem cell factor (40 ng/mL), Flt3L (30 ng/mL), interleukin-3 (IL-3) (10 ng/mL), IL-6 (10 ng/mL), and IL-7 (10 ng/mL; PeproTech, Rocky Hill, NJ). Granulocyte colony-stimulating factor–mobilized peripheral blood normal CD34+ cells were obtained from the Bone Marrow Transplantation Unit, Thomas Jefferson University, and cultured in StemSpan SFEM supplemented with StemSpan CC100 (Stemcell Technologies).
PROTAC synthesis
The supplemental Material provides details on the experimental synthesis of YX-2-107.
Animals
For leukemogenesis assays, 2 × 106 shCDK6-transduced BV173 cells or primary Ph+ ALL cells were injected intravenously into 7- to 9-week-old NOD/SCID/IL-2Rγnull or NRG-SGM3 mice (The Jackson Laboratory, Bar Harbor, ME).
Palbociclib isethionate was purchased from LC Laboratories (Woburn, MA) and mixed in the chow by Research Diets Inc. (New Brunswick, NJ) at 800 mg/kg. This dosing was based on the average daily food intake of NSG mice to deliver 150 mg/kg per day of palbociclib. Palbociclib chow was given ad libitum and replaced every 7 days for the duration of the experiment.
Peripheral blood or bone marrow leukemic cells were detected by anti-human CD19 or CD10 (BD Biosciences, Franklin Lakes, NJ) flow cytometry.
Statistical analyses
Data, expressed as the mean ± standard deviation of 3 experiments, were analyzed for statistical significance by using the unpaired, 2-tailed Student t test. Kaplan-Meier plots of mice survivals were generated by using Prism 6.0 software (GraphPad Software, La Jolla, CA). Differences in survival were assessed by using the log-rank test.
Messenger RNA level correlations were analyzed by using the Pearson test, and significance was calculated by the Student t distribution and corrected for multiple comparisons according to the Bonferroni method.
Results
CDK6 silencing is more effective than CDK4/6 enzymatic inhibition in suppressing Ph+ ALL in immunodeficient mice
Proliferation, CDK4/6–dependent RB phosphorylation and FOXM1 expression18,26 are markedly reduced in CDK6-silenced Ph+ ALL cell lines.6 However, it is unknown whether CDK6 silencing suppresses Ph+ ALL in mice and whether the effects are comparable or superior to CDK4/6 enzymatic inhibition.
First, we determined that CDK6 silencing in Ph+ BV173 cells induced a marked increase in apoptosis that was not detected after treatment with the CDK4/6 inhibitor palbociclib or after CDK4 silencing (Figure 1A-C). To assess if apoptosis induced by CDK6 silencing was specific, shCDK6-88 was cloned in the tetracycline-regulated Tet-pLKO-puro vector and transduced into BV173 cells, which were subsequently transduced with either a vector expressing a CDK6 complementary DNA engineered to prevent the binding of the short hairpin RNA (shRNA) or with the empty vector. Expression of the shRNA-resistant form of CDK6 rescued the apoptosis induced by CDK6 knockdown in shCDK6-BV173 cells, whereas treatment with palbociclib had similarly modest effects in both lines (Figure 1D). Expression of the shRNA-resistant CDK6 also rescued the doxycycline (DOX)-induced decrease in CDK6 and phospho-RB levels, and in the number of S-phase cells (supplemental Figure 1A-B).
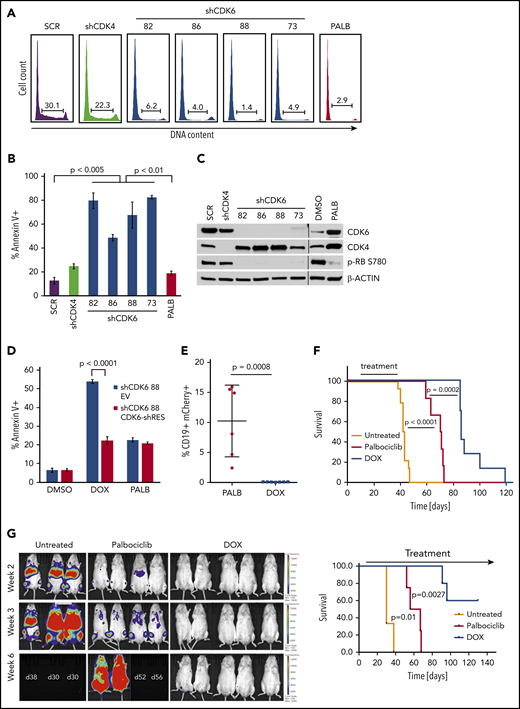
Effect of CDK6 silencing on apoptosis and leukemogenesis of BV173 cells. BV173 cells were transduced with scramble (SCR), CDK4, or CDK6 (82, 86, 88, 73) shRNA vectors and selected with puromycin or treated with palbociclib (2 µM). (A) Cell cycle analysis by propidium iodide staining of shRNA-transduced or palbociclib-treated cells. (B) Apoptosis detected by Annexin V staining after 7 days of puromycin or palbociclib treatment. (C) Representative immunoblot for CDK4/6 and phospho-RB expression. (D) Apoptosis detected by Annexin V staining of BV173 cells transduced with TET-ON shCDK6-88 and treated with DOX (1 µg/mL) or palbociclib (1 µM) for 7 days. (E) Leukemia load (peripheral blood flow cytometry analysis of CD19+mCherry+ cells performed 2 weeks after treatment cessation) of NSG mice injected with BV173 TET-ON shCDK6-88 cells and left untreated or treated with DOX (2 g/L in the drinking water) or palbociclib chow for 4 weeks starting 7 days post–cell injection. (F) Kaplan-Meier survival plot of NSG mice injected with BV173 TET-ON shCDK6-88 and left untreated or treated with DOX (2 g/L in the drinking water) or palbociclib chow for 4 weeks starting 7 days post–cell injection. (G) Serial bioluminescence images (left) and survival (right) of NSG mice injected with shCDK6-BV173-Luc cells and treated continuously with DOX in the drinking water or palbociclib chow.
Effect of CDK6 silencing on apoptosis and leukemogenesis of BV173 cells. BV173 cells were transduced with scramble (SCR), CDK4, or CDK6 (82, 86, 88, 73) shRNA vectors and selected with puromycin or treated with palbociclib (2 µM). (A) Cell cycle analysis by propidium iodide staining of shRNA-transduced or palbociclib-treated cells. (B) Apoptosis detected by Annexin V staining after 7 days of puromycin or palbociclib treatment. (C) Representative immunoblot for CDK4/6 and phospho-RB expression. (D) Apoptosis detected by Annexin V staining of BV173 cells transduced with TET-ON shCDK6-88 and treated with DOX (1 µg/mL) or palbociclib (1 µM) for 7 days. (E) Leukemia load (peripheral blood flow cytometry analysis of CD19+mCherry+ cells performed 2 weeks after treatment cessation) of NSG mice injected with BV173 TET-ON shCDK6-88 cells and left untreated or treated with DOX (2 g/L in the drinking water) or palbociclib chow for 4 weeks starting 7 days post–cell injection. (F) Kaplan-Meier survival plot of NSG mice injected with BV173 TET-ON shCDK6-88 and left untreated or treated with DOX (2 g/L in the drinking water) or palbociclib chow for 4 weeks starting 7 days post–cell injection. (G) Serial bioluminescence images (left) and survival (right) of NSG mice injected with shCDK6-BV173-Luc cells and treated continuously with DOX in the drinking water or palbociclib chow.
Because CDK6 regulates the activity of p53 by enhancing the transcription of p53 antagonists,14 we assessed whether p53 had any role in the apoptosis induced by CDK6 silencing. Downregulation of p53 expression rescued only modestly, albeit significantly, the apoptosis induced by CDK6 silencing (supplemental Figure 1C), indicating that in Ph+ ALL cells apoptosis induced by CDK6 silencing is predominantly p53 independent.
We then assessed whether CDK6 silencing suppressed in vivo growth of Ph+ BV173 cells. To this end, NSG mice were injected with DOX-inducible shCDK6-BV173 cells and left untreated or, 7 days after cell injection, treated with DOX in the drinking water or palbociclib given in the chow to compare the effects of CDK6 silencing vs CDK6 enzymatic inhibition. Treatments were terminated after 4 weeks; 2 weeks later, we assessed the percentage of CD19+ leukemic cells by using peripheral blood flow cytometry. Control mice were all dead at the time of the analysis. CD19+ leukemic cells were barely detectable in DOX-treated mice whereas they comprised ∼10% (range, 2.4%-16%) of total white blood cells in palbociclib-treated animals (Figure 1E), indicating that leukemia load was markedly suppressed by CDK6 silencing.
These data on leukemia burden correlated with markedly different survival of DOX-treated and palbociclib-treated mice. Compared with untreated mice (median survival, 42.5 days), survival of palbociclib-treated mice was significantly prolonged (median survival, 70.5 days; P < .001). However, mice treated with DOX to silence CDK6 expression survived even longer (median survival, 86 days; P = .0002) than palbociclib-treated mice (Figure 1F). In a separate experiment, NSG mice were injected with shCDK6-BV173-Luc cells, and the effects of prolonged CDK6 silencing or palbociclib treatment on disease progression were assessed by bioimaging. Figure 1G shows that CDK6 silencing was markedly more effective than CDK4/6 inhibition in suppressing Ph+ ALL in vivo. Thus, selective CDK6 silencing provides a significant advantage over nonselective CDK4/6 inhibitors such as palbociclib.
The growth suppression induced by CDK6 silencing correlates with a specific gene expression signature
The longer survival of DOX-treated mice compared with palbociclib-treated mice injected with shCDK6-BV173 cells suggests that kinase-independent effects were involved in the more pronounced leukemia suppression induced by CDK6 silencing. To search for kinase-independent pathways potentially explaining this effect, we compared the gene expression profile of untreated (dimethyl sulfoxide [DMSO] control), palbociclib-treated, and CDK6-silenced (DOX-treated; 48 hours) BV173 cells.
As expected, most genes were similarly regulated by CDK6 enzymatic inhibition and silencing; however, 80 genes exhibited at least a 1.5-fold change in expression in CDK6-silenced cells compared with palbociclib-treated cells (Figure 2A). This “CDK6-silencing signature” was obtained by identifying genes upregulated or downregulated by CDK6 silencing only compared with those expressed at similar levels in palbociclib-treated and untreated (DMSO only) BV173 cells. Accordingly, it is highly unlikely that potential changes in gene expression induced by inhibition of CDK4 kinase activity in the palbociclib-treated samples might have inadvertently contributed to the CDK6 silencing signature because these genes would have been excluded by comparing the gene expression profile of palbociclib-treated and control BV173 cells.
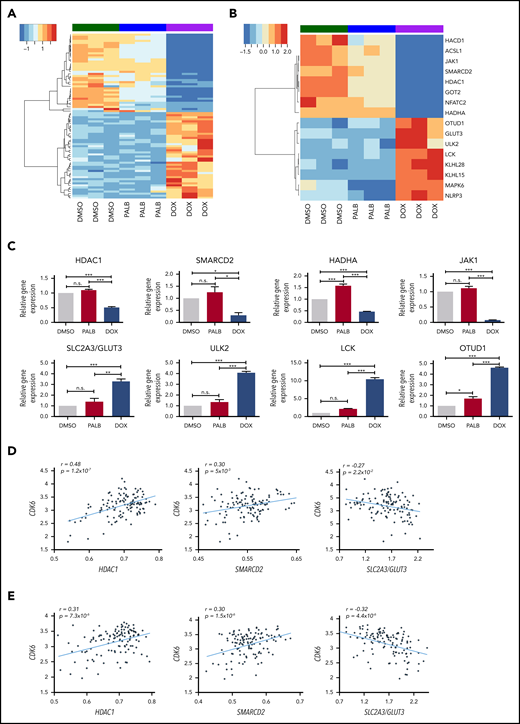
Gene subset regulated by CDK6 silencing not by kinase inhibition in BV173 cells. (A) Heatmap of 80 genes selectively regulated by CDK6 silencing compared with palbociclib treatment in Ph+ BV173 cells. (B) Heatmap of 16 genes selectively downregulated or upregulated by CDK6 silencing. (C) Quantitative polymerase chain reaction analysis of selected genes differentially regulated by CDK6 silencing but not palbociclib treatment in Ph+ BV173 cells. Data represent mean ± standard deviation of 3 independent experiments. Statistical analysis: one-way analysis of variance with Bonferroni’s correction. *P < .05, **P < .01, ***P < .001. Plots of the correlation between the expression of CDK6 and HDAC1, CDK6 and SMARCD2, and CDK6 and SLC2A3/GLUT3 in a panel of 122 Ph+ ALL samples (GSE13159; MILE) (D) and Ph– B-ALL samples (GSE13159;MILE) (E). n.s., not significant.
Gene subset regulated by CDK6 silencing not by kinase inhibition in BV173 cells. (A) Heatmap of 80 genes selectively regulated by CDK6 silencing compared with palbociclib treatment in Ph+ BV173 cells. (B) Heatmap of 16 genes selectively downregulated or upregulated by CDK6 silencing. (C) Quantitative polymerase chain reaction analysis of selected genes differentially regulated by CDK6 silencing but not palbociclib treatment in Ph+ BV173 cells. Data represent mean ± standard deviation of 3 independent experiments. Statistical analysis: one-way analysis of variance with Bonferroni’s correction. *P < .05, **P < .01, ***P < .001. Plots of the correlation between the expression of CDK6 and HDAC1, CDK6 and SMARCD2, and CDK6 and SLC2A3/GLUT3 in a panel of 122 Ph+ ALL samples (GSE13159; MILE) (D) and Ph– B-ALL samples (GSE13159;MILE) (E). n.s., not significant.
Among genes selectively downregulated by CDK6 silencing, JAK1 and NFATC2 could affect prosurvival signaling, whereas lower levels of SMARCD2 and HDAC1 could influence chromatin remodeling, possibly affecting transcription of prosurvival genes (Figure 2B-C).
CDK6 silencing may also affect fatty acid metabolism. This possibility is suggested by decreased expression of the HACD1, ACSL1, and HADHA genes and oxidative phosphorylation based on decreased expression of the GOT2 gene, which encodes for the mithocondrial aspartate aminotransferase protein, a component of the malate-aspartate shuttle responsible for the transfer of NADPH reducing equivalents across the mithocondrial inner membrane.
Among genes selectively upregulated by CDK6 silencing (Figure 2B-C), SLC2A3/GLUT3 may promote glucose transport, enhancing glycolytic ATP production, and ULK2 and LCK1 may induce a state of autophagy and an anergic phenotype (unresponsiveness to BCR stimulation).27 OTUD1 encodes for a protein with deubiquitinase activity that negatively regulates the nuclear transport of the growth-promoting YAP1 transcription factor,28 whereas KLHL15 and KLHL28 encode for Kelch-like proteins that contain the BTB (POZ) transcription repressor domain.
To further investigate whether the CDK6-silencing signature might be clinically significant, messenger RNA levels of CDK6 and its putative targets were analyzed in a dataset of 122 Ph+ ALL samples collected from 8 laboratories participating in the MILE (Microarray Innovations in Leukemia) study.29,30 We observed a strikingly positive correlation between CDK6 and HDAC1 expression (P = 1.2 ×10−7), a significant positive correlation between CDK6 and SMARCD2 expression (P = 5 × 10−3), and a significant negative correlation between CDK6 and SLC2A3/GLUT3 (P = 2.2 ×10−2) (Figure 2D).
Development of a potent CDK4/6–targeted PROTAC that selectively degrades CDK6
Based on Figure 1 ex vivo and in vivo data, a compound selectively degrading CDK6 should be more effective than a CDK4/6 enzymatic inhibitor in exploiting the CDK6 dependence of Ph+ ALL.
Conceivably, a potent CDK4/6 kinase inhibitor tethered to an E3 ligase–recruiting molecule might preferentially bind to and degrade CDK6, potentially providing more specific and durable inhibition than currently possible with small molecule inhibitors. We therefore designed CDK4/6–targeted PROTACs guided by the X-ray crystal structure of palbociclib in complex with CDK6 (PDB identifiers, 2EUF and 5L2T).31,32 This structure shows that the piperazine tail of palbociclib protrudes from the CDK6-active site (ATP pocket) toward the solvent, suggesting that it might tolerate linker attachment to various E3 ligase–recruiting molecules.31 First, we synthesized palbociclib derivatives with various linkers; a small representative set is shown in Figure 3A. The kinase inhibitory activity of these conjugates varied dramatically, which was surprising based on the seemingly large volume of solvent-exposed space suggested by the crystal structure in the region where the piperazine tail conjugated with a linker would exit into the solvent. Notably, a single methyl group change in YX-2-11533 compared with AC-1-079 resulted in a 100-fold decrease in kinase activity inhibition. Figure 3B shows several PROTACs that have different linkers and either a von Hippel-Lindau– or cereblon-recruiting ligand.34,35 These compounds were weak inhibitors in in vitro assays of cyclin D3/CDK6– or cyclin D1/CDK4–dependent RB phosphorylation. By contrast, the cereblon-recruiting PROTAC YX-2-107 was a potent inhibitor of in vitro CDK4 or CDK6 kinase activity (50% inhibitory concentration [IC50] = 0.69 and 4.4 nM, respectively) (Figure 3C), comparable to that of palbociclib (IC50 = 11 and 9.5 nM) and a selective CDK6 degrader in Ph+ BV173 ALL cells with a degradation constant of ∼4 nM, based on densitometric analysis of CDK6 band intensity.
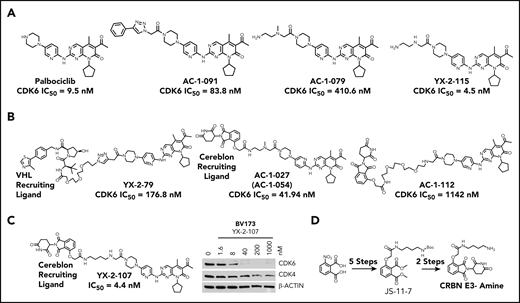
Palbociclib and derivatives. (A) Palbociclib and derivative compounds with differences in kinase inhibition due to modest changes to the piperazine-linker tail. (B) Several PROTAC candidates using various linkers and either a von Hippel-Lindau (VHL)- or a cereblon-recruiting ligand. (C) YX-2-107, a CRBN-palbociclib PROTAC, selectively degrades CDK6 in BV173 cells after 4 hours of treatment. (D) Synthesis of CRBN E3-amine component for cereblon E3 ligase recruitment and as a control.
Palbociclib and derivatives. (A) Palbociclib and derivative compounds with differences in kinase inhibition due to modest changes to the piperazine-linker tail. (B) Several PROTAC candidates using various linkers and either a von Hippel-Lindau (VHL)- or a cereblon-recruiting ligand. (C) YX-2-107, a CRBN-palbociclib PROTAC, selectively degrades CDK6 in BV173 cells after 4 hours of treatment. (D) Synthesis of CRBN E3-amine component for cereblon E3 ligase recruitment and as a control.
Figure 3D shows the synthesis of CRBN E3 amine that serves as a recruiter for cereblon E3 ligase. Schematic steps for PROTAC YX-2-107 synthesis are presented in supplemental Figure 2.
In BV173 cells, decreased CDK6 expression was detected as early as 1 hour post-treatment with YX-2-107; up to 12 hours, CDK6 levels were markedly reduced (supplemental Figure 3A-B). At 24 hours post-treatment, CDK6 expression was still lower than in untreated cells, returning to such levels only after 36 hours of treatment (supplemental Figure 3B). The rapid degradation of CDK6 induced by YX-2-107 was blocked by treatment with the proteasome inhibitor MG132 (supplemental Figure 3C), strongly suggesting that, in YX-2-107–treated BV173 cells, CDK6 is targeted for proteasome degradation via cereblon-dependent ubiquitination. Cotreatment with palbociclib or thalidomide to block binding of PROTAC YX-2-107 to CDK6 or cereblon, respectively, prevented the downregulation of CDK6 (supplemental Figure 3D). This finding suggests that YX-2-107–induced CDK6 degradation requires the formation of a ternary complex consisting of CDK6 + PROTAC + cereblon. Expression of CDK6 in PROTAC YX-2-107–treated BV173 cells was very low for at least 6 hours, returning to the levels of untreated cells 12 hours after washing the PROTAC from the culture medium (supplemental Figure 3E); this finding suggests that the inhibitory effect for this PROTAC is more durable than for the CDK4/6 inhibitor palbociclib.
Proteomic analysis of YX-2-107–treated (4 hours) BV173 cells revealed that of 3682 proteins examined, only CDK6 was significantly downregulated, compared with control (DMSO-treated) cells (supplemental Figure 3F). Interestingly, expression of IKZF1 and IKZF3, which was destabilized by a CDK4/6–targeted pomalidomide-based PROTAC,36 was not affected by treatment of BV173 cells with YX-2-107.
Because CDK4 is exclusively localized in the cytoplasm of Ph+ ALL cells whereas CDK6 is predominantly nuclear,6 we sought to determine whether this finding might explain the preferential CDK6 degradation by cereblon, which is, in part, localized in the nucleus.37 Thus, BV173 cells expressing a nuclearly localized CDK4 protein (NLS-CDK4-BV173)6 were treated with PROTAC YX-2-107, and levels of NLS-CDK4 were assessed by western blotting. NLS-CDK4 was not degraded by PROTAC YX-2-107 (supplemental Figure 4), strongly suggesting that the nuclear localization of CDK6 as opposed to CDK4 in Ph+ ALL cells does not account for the preferential degradation of CDK6 over CDK4 by PROTAC YX-2-107.
In contrast, PROTAC YX-2-233, a palbociclib derivative conjugated to an MDM2-recruiting ligand derived from RG7112,38 potently suppressed S phase and RB phosphorylation in Ph+ ALL cells and degraded CDK4 and CDK6 (supplemental Figure 5). These findings suggest that the E3 ligase recruited by the PROTAC may influence the selective degradation of a targeted protein.
Effects of PROTAC YX-2-107 in Ph+ ALL cell lines and normal hematopoietic progenitors
Treatment with YX-2-107 inhibited S-phase entry, cell proliferation, RB phosphorylation, and FOXM1 expression and induced the selective degradation of CDK6 in Ph+ BV173 and SUP-B15 cells (Figure 4A-F); treatment with palbociclib induced increased expression of CDK4 and, especially, CDK6. The control compound, CRBN E3-Amine (CRBN-L) (Figure 3D) that consists exclusively of the cereblon E3 ligase–recruiting ligand, had no effect on the percentage of S-phase cells or CDK4/6, phospho-RB, and FOXM1 expression (Figure 4A-D).
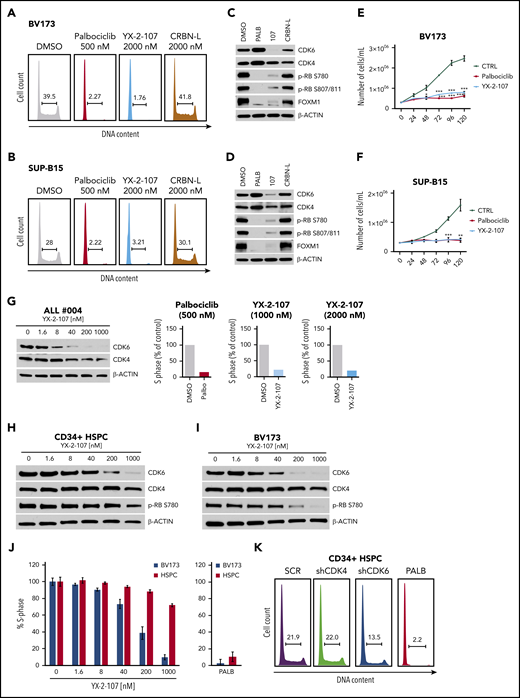
Effects of PROTAC YX-2-107 in Ph+ ALL cells and normal hematopoietic progenitors. Cell cycle analysis of BV173 cells (A) and SUP-B15 cells (B) after 48 hours of treatment with the indicated drug concentrations. Immunoblot of BV173 cells (C) and SUP-B15 cells (D) showing the expression of CDK6, CDK4, FOXM1, and phospho-RB after 72 hours of treatment with the indicated drug concentrations. Cell counts (Trypan blue staining) (mean ± standard deviation; 3 independent experiments) of palbociclib-treated and PROTAC YX-2-107–treated (1 µM added each day) BV173 (E) or SUP-B15 (F) cells. (G) Immunoblot for CDK4/CDK6 expression (left) and number of S-phase cells (represented as the percentage of drug-treated vs untreated cells taken as 100) (right) in YX-2-107–treated Ph+ ALL cells (sample #004). (H-J) Immunoblot for CDK4/CDK6 expression and percentage of S-phase cells in YX-2-107–treated normal hematopoietic progenitors and BV173 cells. (K) Cell cycle profile of CD34+ hematopoietic stem and progenitor cells (HSPC) transduced with anti-CDK4 or anti-CDK6 shRNA and selected with puromycin for 48 hours or treated with palbociclib (500 nM; 24 hours).
Effects of PROTAC YX-2-107 in Ph+ ALL cells and normal hematopoietic progenitors. Cell cycle analysis of BV173 cells (A) and SUP-B15 cells (B) after 48 hours of treatment with the indicated drug concentrations. Immunoblot of BV173 cells (C) and SUP-B15 cells (D) showing the expression of CDK6, CDK4, FOXM1, and phospho-RB after 72 hours of treatment with the indicated drug concentrations. Cell counts (Trypan blue staining) (mean ± standard deviation; 3 independent experiments) of palbociclib-treated and PROTAC YX-2-107–treated (1 µM added each day) BV173 (E) or SUP-B15 (F) cells. (G) Immunoblot for CDK4/CDK6 expression (left) and number of S-phase cells (represented as the percentage of drug-treated vs untreated cells taken as 100) (right) in YX-2-107–treated Ph+ ALL cells (sample #004). (H-J) Immunoblot for CDK4/CDK6 expression and percentage of S-phase cells in YX-2-107–treated normal hematopoietic progenitors and BV173 cells. (K) Cell cycle profile of CD34+ hematopoietic stem and progenitor cells (HSPC) transduced with anti-CDK4 or anti-CDK6 shRNA and selected with puromycin for 48 hours or treated with palbociclib (500 nM; 24 hours).
Selective degradation of CDK6 and inhibition of S phase (DNA content analysis and 5-ethynyl-2′-deoxyuridine labeling) was also observed in YX-2-107–treated blasts from a patient with de novo Ph+ ALL (Figure 4G).
Inhibition of S phase and selective degradation of CDK6 was also detected in YX-2-107–treated Ph-like (MUTZ-5 and MHH-CALL-4) and MLL-rearranged (SEM) cell lines but not in the B-ALL line 697 (1;19, translocation) (supplemental Figure 6A). Of interest, YX-2-107 treatment of the Jurkat T-ALL line led to a marked decrease in CDK6 levels with no effects on CDK4 expression; however, S phase was not inhibited in this cell line, suggesting that either CDK4 activity compensates for CDK6 loss or that this leukemia line is CDK4 dependent. It is noteworthy that expression of CDK6 in the adult Ph-like and MLL-rearranged B-ALL is comparable/higher than in Ph+ ALL (supplemental Figure 6B), confirming findings from RNA-sequencing analyses of a recently published study of B-progenitor acute lymphoblastic leukemia subsets.39
Decreased CDK6, but not CDK4 expression, was also observed in YX-2-107–treated normal CD34+ progenitors; however, this treatment did not inhibit S phase or reduce phospho-RB as effectively as in BV173 cells (Figure 4H-J). By contrast, treatment with the CDK4/6 inhibitor palbociclib had similar growth-suppressive effects in BV173 and normal CD34+ cells, suggesting that, unlike Ph+ ALL cells, normal CD34+ cells rely on both CDK4 and CDK6 for their growth. This hypothesis was confirmed by the observation that silencing CDK4 alone did not decrease the percentage of CD34+ S-phase cells, whereas selective CDK6 silencing had only a partial effect compared with treatment with palbociclib, which instead markedly suppressed CD34+ S phase cells (Figure 4K).
Additional PROTACs (AC-2-011, AC-1-212, and AC-1-277) (supplemental Figure 7A) inhibited RB phosphorylation and markedly reduced S phase in BV173 cells; however, AC-2-011 is not a potent CDK6 degrader, as it degraded CDK6 and CDK4 at high concentrations (supplemental Figure 7B-C). By contrast, AC-1-212 and AC-1-277 degraded CDK6 selectively and potently, and suppressed the number of BV173 S phase cells with similar or higher potency as palbociclib.
PROTAC YX-2-107 is bioavailable in mice and pharmacologically active in suppressing Ph+ ALL proliferation
To assess the potential use of CDK6-selective PROTACs as drugs in vivo, we evaluated the metabolic stability of YX-2-107 in mouse liver microsomes and compared it to palbociclib and to 4-hydroxy-thalidomide (AC-1-158) (Figure 5A). YX-2-107 has good metabolic stability, displaying a half-life of 35 minutes, compared with palbociclib, which exhibited a half-life of 56 minutes. Control compound midazolam (poor stability) had a half-life of ∼2 minutes. Other derivatives showed poor (ie, AC-1-027; 2-minute half-life) (Figure 3B) or moderate (ie, AC-1-212; 10-minute half-life) stability (supplemental Figure 7A), emphasizing the importance of compound optimization before in vivo studies. We next evaluated YX-2-107 in a mouse pharmacokinetic (PK) study at a 10 mg/kg intraperitoneal dose (Figure 5B). Plasma levels show a maximum concentration of 741 nM (150-fold greater than CDK6 degradation IC50), with clearance from the plasma after 4 hours. The plasma exposure is 30-fold higher than CDK6 degradation IC50 at 2 hours (133 nM) and ∼4-fold higher at 6 hours (21 nM). Conceivably, CDK6 inhibition may persist for an extended time period beyond clearance of YX-2-107 based on the recovery time of CDK6 de novo protein synthesis in PROTAC YX-2-107–treated BV173 cells (supplemental Figure 3D), providing an advantage over palbociclib or other ATP competitive inhibitors. Although the PK profile of PROTAC YX-2-107 may not be optimal for a clinical compound, it is suitable for a feasibility study to evaluate its growth-suppressive effects in vivo.
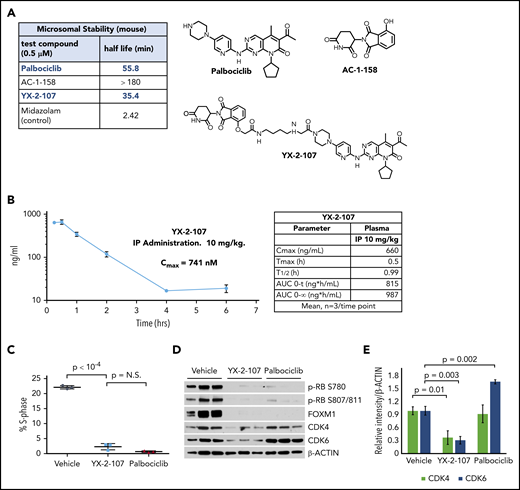
PROTAC YX-2-107 metabolic stability and its biological activity in a mouse xenograft of Ph+ ALL. (A) Half-life of YX-2-107, palbociclib, and E3 ligase–recruiting molecules incubated in mouse liver microsomes. (B) Time course of plasma concentration of YX-2-107 injected intraperitoneally at 10 mg/kg into C57BL/6j mice and its PK property (left). Cell cycle analysis by propidium iodide staining (C) and immunoblot for phospho-RB, FOXM1, CDK4, and CDK6 (D), with densitometry of CDK4 and CDK6 expression (E) of bone marrow leukemic cells (>90% CD19+CD10+ by flow cytometry), from NSG mice injected with Ph+ ALL cells and treated (3 mice/group) with palbociclib or YX-2-107 at 150 mg/kg per day for 3 consecutive days when peripheral blood leukemic cells were 50%. Bone marrow leukemic cells were purified 24 hours after the cessation of drug treatment. N.S., not significant.
PROTAC YX-2-107 metabolic stability and its biological activity in a mouse xenograft of Ph+ ALL. (A) Half-life of YX-2-107, palbociclib, and E3 ligase–recruiting molecules incubated in mouse liver microsomes. (B) Time course of plasma concentration of YX-2-107 injected intraperitoneally at 10 mg/kg into C57BL/6j mice and its PK property (left). Cell cycle analysis by propidium iodide staining (C) and immunoblot for phospho-RB, FOXM1, CDK4, and CDK6 (D), with densitometry of CDK4 and CDK6 expression (E) of bone marrow leukemic cells (>90% CD19+CD10+ by flow cytometry), from NSG mice injected with Ph+ ALL cells and treated (3 mice/group) with palbociclib or YX-2-107 at 150 mg/kg per day for 3 consecutive days when peripheral blood leukemic cells were 50%. Bone marrow leukemic cells were purified 24 hours after the cessation of drug treatment. N.S., not significant.
Thus, we tested the in vivo effects of PROTAC YX-2-107 after a short treatment in Ph+ ALL xenografts. For this experiment, mice (n = 9; three/group) were injected with primary Ph+ ALL cells, monitored for the presence of leukemic cells (CD19+CD10+) in the peripheral blood, and treated (3 consecutive days) with palbociclib, YX-2-107, or vehicle only when these cells were >50%. Subsequently, bone marrow cells (>90% CD19+/CD10+) were purified and assessed for cell cycle activity, phospho-RB, FOXM1, and CDK4/CDK6 levels. Palbociclib and YX-2-107 were indistinguishable in suppressing S-phase cell percentage (Figure 5C) and decreasing phospho-RB and FOXM1 expression (Figure 5D). However, treatment with PROTAC YX-2-107 reduced CDK6 and, to a lesser degree, CDK4 levels; conversely, CDK6 expression was upregulated by palbociclib (Figure 5D-E). A similar pilot study was performed with PROTAC AC-1-212. Treatment with this PROTAC suppressed the percentage of primary Ph+ ALL S-phase cells, the expression of CDK4/6–regulated phospho-RB and, to a lesser degree, FOXM1, and induced the selective CDK6 degradation (supplemental Figure 8). However, AC-1-212 was less effective than YX-2-107, and the effects were not dose-dependent (compare Figure 5C and supplemental Figure 8A; data not shown), probably reflecting its suboptimal stability (10-minute half-life) in mouse liver microsomes.
Effects of CDK6-degrader PROTAC YX-2-107 in patient-derived xenografts of Ph+ ALL
Based on the in vivo data with YX-2-107, we further investigated its effects in Ph+ leukemia xenografts. First, we assessed whether in vivo treatment with YX-2-107 was toxic for normal hematopoietic cells. To this end, six C57BL/6j mice were treated with YX-2-107 at a dose of 150 mg/kg for 10 consecutive days. The mice did not display signs of distress or loss of weight during the treatment (not shown). Four days after termination of the treatment, the mice were euthanized, and peripheral blood and bone marrow cells were purified. Flow cytometry analysis of bone marrow cells found no significant changes in the percentage of stem and progenitor cells and of B-cell precursors. Likewise, counts of peripheral blood cell subsets were normal except for a moderate increase in platelets and reticulocytes (Figure 6).
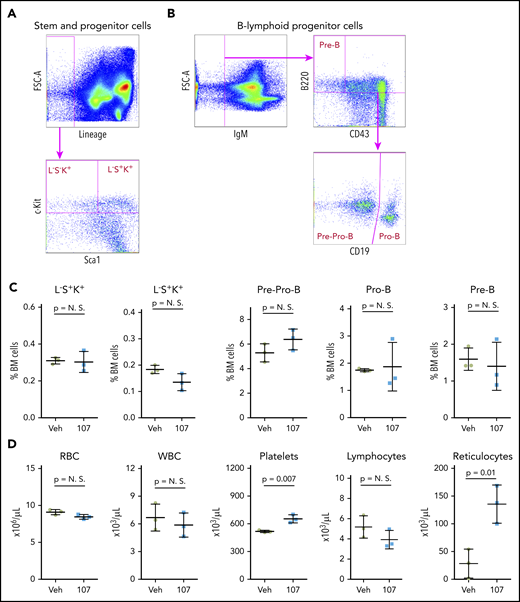
Effect of PROTAC YX-2-107 treatment on normal mouse hematopoiesis. Six (2 month-old) C57BL/6j mice were treated with vehicle (Veh) or PROTAC YX-2-107 (107) 150 mg/kg intraperitoneally daily for 10 consecutive days. Four days after cessation of treatment, peripheral blood and bone marrow (BM) cells were collected and analyzed by flow cytometry. (A) Gating strategy for the quantification of stem and progenitor cells. (B) Gating strategy for the quantification of B-lymphoid progenitor cells. (C) Percentage of progenitor populations in the BM. (D) Number of selected hematopoietic cells in the peripheral blood. P value was considered nonsignificant (N.S.) if >.05.
Effect of PROTAC YX-2-107 treatment on normal mouse hematopoiesis. Six (2 month-old) C57BL/6j mice were treated with vehicle (Veh) or PROTAC YX-2-107 (107) 150 mg/kg intraperitoneally daily for 10 consecutive days. Four days after cessation of treatment, peripheral blood and bone marrow (BM) cells were collected and analyzed by flow cytometry. (A) Gating strategy for the quantification of stem and progenitor cells. (B) Gating strategy for the quantification of B-lymphoid progenitor cells. (C) Percentage of progenitor populations in the BM. (D) Number of selected hematopoietic cells in the peripheral blood. P value was considered nonsignificant (N.S.) if >.05.
Because such treatment was well tolerated by normal mice, we compared the effects of palbociclib and YX-2-107 on peripheral blood leukemia load (percentage of CD19+CD10+ cells) in NSG mice injected with primary Ph+ ALL cells from 2 patients and treated for 10 consecutive days with palbociclib or YX-2-107 (125 or 150 mg/kg intraperitoneally; once a day or twice daily at half-dose per injection). Treatment with YX-2-107 was as effective as palbociclib in suppressing peripheral blood leukemia burden (supplemental Figure 9). The twice-daily treatment with YX-2-107 seemed to be more effective than the single-dose per day treatment regimen, at least in mice injected with ALL sample #1222, which is consistent with YX-2-107 plasma exposure in which the compound is cleared after ∼4 hours. However, leukemia growth resumed rapidly upon cessation of treatment with either drug (not shown).
Lastly, we assessed the peripheral blood leukemia load of NRG-SGM3 mice injected with a TKI-resistant primary Ph+ ALL sample and treated for 20 consecutive days with palbociclib or YX-2-107 (25 mg or 50 mg/kg twice per day). As shown in Figure 7, YX-2-107 seems to be more effective than palbociclib in suppressing the in vivo growth of this TKI-resistant Ph+ ALL.
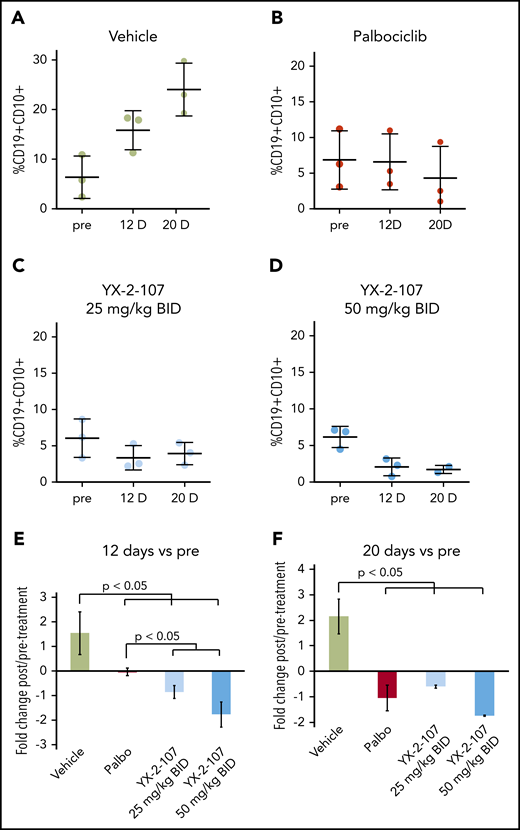
Effect of PROTAC YX-2-107 on peripheral blood leukemia burden of NSG mice injected with a TKI-resistant Ph+ ALL. NSG mice were injected with a primary, human, TKI-resistant (BCR-ABL1T315I) Ph+ ALL sample (#557). (A-D) Peripheral blood leukemia burden was analyzed at week 7 following cell injection (pre), and after 12 and 20 days of treatment with palbociclib (mixed in the diet to achieve a dose of 150 mg/kg per day) or YX-2-107 intraperitoneally (in a Kolliphor/PBS/DMSO 20:70:10 suspension, twice daily at either 25 mg/kg or 50 mg/kg). (E-F) Fold changes of the percentages shown above.
Effect of PROTAC YX-2-107 on peripheral blood leukemia burden of NSG mice injected with a TKI-resistant Ph+ ALL. NSG mice were injected with a primary, human, TKI-resistant (BCR-ABL1T315I) Ph+ ALL sample (#557). (A-D) Peripheral blood leukemia burden was analyzed at week 7 following cell injection (pre), and after 12 and 20 days of treatment with palbociclib (mixed in the diet to achieve a dose of 150 mg/kg per day) or YX-2-107 intraperitoneally (in a Kolliphor/PBS/DMSO 20:70:10 suspension, twice daily at either 25 mg/kg or 50 mg/kg). (E-F) Fold changes of the percentages shown above.
Discussion
We report the development of selective CDK6-degrading PROTACs consisting of high-affinity small molecule ligands for CDK4/6, and for the E3 ubiquitin ligase cereblon, joined by linkers of different structure and/or size. By contrast, PROTAC YX-2-233, which uses as the E3 ubiquitin ligase recruiter the MDM2 ligand RG7112,38 degraded CDK4 and CDK6 with equal efficiency. The selective degradation of CDK6 by cereblon-recruiting PROTACs may be explained by formation of a ternary complex that generates new protein–protein contacts, allowing selective lysine ubiquitination of CDK6 over CDK4, followed by 26S proteasomal degradation. This explanation is consistent with recent findings showing that a CDK4/6–targeted cereblon-recruiting PROTAC induced in live cells the formation of a ternary complex with CDK6 and cereblon but not with CDK4 and cereblon.36
Selective degraders of CDK6 are attractive therapeutic agents in Ph+ ALL because Ph+ ALL cells require CDK6, but not CDK4, expression for their growth,6 and CDK6 has kinase-independent growth-promoting effects8-13 that can be exploited therapeutically only by CDK6-degrading drugs. Indeed, CDK6 silencing was more effective than palbociclib treatment in suppressing Ph+ ALL in vivo, possibly due to reduced expression of genes involved in cell survival, chromatin remodeling, and mitochondrial metabolic pathways (Figure 2).
Interestingly, CDK6 and HDAC1 expression was highly correlated in a dataset of Ph+ and Ph– ALL patient samples (Figure 2D), suggesting that reduced HDAC1 levels may be critical for the growth suppression/apoptosis of CDK6-silenced Ph+ ALL cells. In this regard, genetic or pharmacologic inhibition of HDAC1 induced apoptosis of several B-ALL lines, although Ph+ cell lines were not among those tested.40
Among our CDK6-degrading PROTACs, we investigated in detail one compound, termed YX-2-107. This compound inhibited proliferation, phospho-RB, FOXM1, and CDK6, but not CDK4, expression in Ph+ ALL cells. Moreover, treatment with YX-2-107 had similar effects in two Ph-like B-ALL cell lines (MUTZ-5 and MHH-CALL-4) and in one MLL-rearranged (SEM) B-ALL line. In contrast, in normal CD34+ hematopoietic progenitors, which depend on the expression/activity of both CDK4 and CDK6 for their proliferation, YX-2-107 induced the selective degradation of CDK6 but did not significantly inhibit the S phase of these cells. These findings imply that, in the clinic, selective CDK6 degraders would be less toxic for normal hematopoietic cells than dual CDK4/6 inhibitors.
Indeed, neutropenia was the most common adverse event (60%-70%) in patients with breast cancer treated with palbociclib or ribociclib.41,42 Such an adverse event would probably be clinically relevant in patients with acute leukemia and low normal white blood cell counts due to bone marrow replacement by leukemic cells, emphasizing the importance of using a selective CDK6 inhibitor to spare normal hematopoietic progenitors.
YX-2-107 was relatively stable when incubated in mouse liver microsomes, but it had a half-life of only 1 hour in a mouse PK study (Figure 5B), suggesting that further improvement in PK parameters is warranted.
Nevertheless, a short-term YX-2-107 treatment of NSG mice with Ph+ ALL significantly inhibited the number of bone marrow S-phase cells, markedly suppressed the expression of CDK4/6 substrates phospho-RB and FOXM1, and induced the preferential degradation of CDK6 over CDK4.
A longer YX-2-107 treatment of mice injected with de novo or TKI-resistant primary Ph+ ALL induced a marked suppression of peripheral blood leukemia load that was comparable or even superior to that induced by palbociclib.
Normal mice treated for 10 consecutive days with a high dose of YX-2-107 did not exhibit significant changes in normal hematopoietic progenitors and mature cells (Figure 6), consistent with the modest effects of YX-2-107 on the S phase of human CD34+ cells (Figure 4).
During the course of these studies, other laboratories reported the synthesis of cereblon-recruiting palbociclib-based CDK6-degrading PROTACs.36,43-45 However, analysis of the biological effects of these compounds was rather limited,36,43,45 and no studies were performed to assess the therapeutic effects of these compounds in tumor mouse models. We expect that further development of YX-2-107 or of other CDK6-selective PROTACs will yield second-generation derivatives combining superior ex vivo activities (inhibition of CDK6-regulated S phase; CDK6 degradation) with improved PK properties. Such drugs could serve as bona fide anticancer therapeutic agents with novel mechanisms of action for Ph+ ALL and other CDK6-dependent malignancies, including Ph-like B-ALL and MLL-rearranged B-ALL.
For materials and methods, please e-mail requests to the corresponding author. RNA sequencing data: BioProject database; submission ID: SUB6392472; BioProject ID: PRJNA576003.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Christine Eischen and Mark Fortini for critical reading of the manuscript.
This work was supported by National Institutes of Health (NIH)/National Cancer Institute grants RO1-CA167169 (B.C), R50 CA221838 (H.Y.-T.), and P30 CA010615 (J.M.S.), and NIH/National Heart, Lung, and Blood Institute grant RO1-HL127895 (B.C. and A.M.). Support for the Flow Cytometry, Cancer Genomics and Bioinformatics, and Laboratory Animal Facility was provided by the Cancer Center Support Grant CA056036 to the Sidney Kimmel Cancer Center at Thomas Jefferson University. Support for the Wistar Proteomics and Metabolomics Core Facility was provided by Cancer Center Support Grant CA010815 to The Wistar Institute.
Authorship
Contribution: M.D.D. contributed to the design of the studies and performed most of the experiments; P.P. performed several experiments, including those suggested by the reviewers; Y.X. and A.C. performed the synthesis of the PROTACs; H.-T.Y. performed the proteomic analysis of PROTAC YX-2-107–treated cells; G.K. and P.F. performed RNA-sequencing and bioinformatics analysis of palbociclib-treated and CDK6-silenced cells; O.S., A.R., and L.F.P. provided the primary Ph+ ALL samples; S.P. performed the 5-ethynyl-2′-deoxyuridine–labeling experiment of PROTAC-treated primary Ph+ ALL cells; C.B. helped P.P. in performing cell counts of PROTAC-treated cells; A.M. and G.C. provided advice on experiments and on the organization of the manuscript; J.M.S. was responsible for the design and synthesis of the PROTACs and control compounds used in the study and for writing parts of the manuscript; and B.C. was responsible for designing most of the experiments and for writing the manuscript.
Conflict-of-interest disclosure: J.M.S. has financial interest in Alliance Discovery, Inc, Context Therapeutics, Inc, and The Barer Institute. The remaining authors declare no competing financial interests.
The current affiliation for M.D.D. is Department of Biochemistry and Molecular Genetics, University of Colorado, Anschutz Medical Campus, Aurora, CO.
Correspondence: Bruno Calabretta, Department of Cancer Biology and Sidney Kimmel Cancer Center, Thomas Jefferson University, 233 South 10th St, Philadelphia, PA 19107; e-mail: bruno.calabretta@jefferson.edu; or Joseph M. Salvino, Molecular and Cellular Oncogenesis Program, The Wistar Institute, 3601 Spruce St, Philadelphia, PA 19104; e-mail: jsalvino@wistar.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal