

Key Points
Chk, Csk, and PTPRJ are the main regulators of SFKs in platelets, which play a key role in maintaining platelet count.
SFKs are able to autoregulate their activity by phosphorylating either their activation loop or C-terminal inhibitory tyrosine residue.
Abstract
The Src family kinases (SFKs) Src, Lyn, and Fyn are essential for platelet activation and also involved in megakaryocyte (MK) development and platelet production. Platelet SFKs are inhibited by C-terminal Src kinase (Csk), which phosphorylates a conserved tyrosine in their C-terminal tail, and are activated by the receptor-type tyrosine phosphatase PTPRJ (CD148, DEP-1), which dephosphorylates the same residue. Deletion of Csk and PTPRJ in the MK lineage in mice results in increased SFK activity, but paradoxically hypoactive platelets resulting from negative feedback mechanisms, including upregulation of Csk homologous kinase (Chk) expression. Here, we investigate the role of Chk in platelets, functional redundancy with Csk, and the physiological consequences of ablating Chk, Csk, and PTPRJ in mice. Platelet count was normal in Chk knockout (KO) mice, reduced by 92% in Chk;Csk double KO (DKO) mice, and partially rescued in Chk;Csk;Ptprj triple KO (TKO) mice. Megakaryocyte numbers were significantly increased in both DKO and TKO mice. Phosphorylation of the inhibitory tyrosine of SFKs was almost completely abolished in DKO platelets, which was partially rescued in Src and Fyn in TKO platelets. This residual phosphorylation was abolished by Src inhibitors, revealing an unexpected mechanism in which SFKs autoinhibit their activity by phosphorylating their C-terminal tyrosine residues. We demonstrate that reduced inhibitory phosphorylation of SFKs leads to thrombocytopenia, with Csk being the dominant inhibitor in platelets and Chk having an auxiliary role. PTPRJ deletion in addition to Chk and Csk ameliorates the extent of thrombocytopenia, suggesting targeting it may have therapeutic benefits in such conditions.

Introduction
Src family kinases (SFKs) are essential for initiating and propagating activation signals from a variety of platelet receptors, including the immunoreceptor tyrosine-based activation motif (ITAM)-containing collagen receptor complex GPVI-FcR γ-chain and the fibrinogen receptor integrin αIIbβ3.1 SFKs also initiate inhibitory signaling from immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing receptors, including the megakaryocyte and platelet inhibitory receptor G6b-B (MPIG6B) and platelet endothelial cell adhesion molecule (PECAM-1).2,3 In platelets, SFKs are regulated by C-terminal Src kinase (Csk), which phosphorylates a conserved tyrosine residue in the C-terminal tail of SFKs, restraining them in an inactive conformation, and by the receptor-type tyrosine phosphatase PTPRJ (CD148, DEP-1, RPTPη), which dephosphorylates the same tyrosine residue, thereby releasing SFKs from their autoinhibited conformation.2,4-6 Trans-auto-phosphorylation of a conserved tyrosine residue in their activation loop stabilizes the active conformation. PTPRJ is also able to attenuate SFK activity by dephosphorylating the activation loop tyrosine.2,6-8 Targeted deletion of Csk and Ptprj in the megakaryocyte (MK) lineage of mice results in significantly elevated SFK activity, but paradoxical hypoactive platelets, reduced thrombosis, and increased bleeding, resulting from negative feedback pathways, including downregulation of GPVI-FcR γ-chain and the hemi-ITAM-containing podoplanin receptor CLEC-2, and concomitant upregulation and phosphorylation of the inhibitory receptor MPIG6B.2
Src and Lyn also play important roles in MK development and platelet production, as exemplified by human and mouse genetic studies. The gain-of-function mutation in human SRC (E527K), resulting in loss of Src autoinhibition, causes thrombocytopenia and a reduction in proplatelet formation.9,10 Moreover, Lyn knockout (KO) mice display increased MK progenitor cell proliferation and a greater number of mature MKs with increased ploidy in vitro and more MKs in the bone marrow.11-13 Although Lyn KO and Src KO mice do not display major differences in platelet count compared with control mice because of the overlapping roles of these SFKs; mice deficient in both Lyn and Src develop thrombocytopenia.14 The critical role of Src and Lyn in MK maturation and proplatelet formation is further consolidated by tyrosine kinase inhibitor studies. Pharmacological inhibition of SFKs by PP1, PP2, SU6656, or dasatinib results in enhanced proliferation and maturation of cultured megakaryocytes11,15,16 and increased platelet production of inhibitor-treated MKs infused to mice in vivo.17
The 2 main regulators of platelet SFKs, namely Csk and PTPRJ, also play key roles in MK function and platelet production. Pf4-Cre-generated Csk KO mice display a 65% reduction in platelet count, whereas Gp1ba-Cre-generated Csk KO mice, in which recombination occurs later in MK development, show a 32% decrease in platelet count, highlighting a key role of Csk in MK development and platelet production.2,18 Ptprj KO mice have normal platelet counts, however MKs from these mice display reduced spreading on collagen-, fibrinogen-, and fibronectin-coated surfaces and are unable to migrate toward an SDF-1α gradient.5 Csk;Ptprj double KO (DKO) mice also present normal mean platelet counts, demonstrating that the deletion of Ptprj rescues the platelet count phenotype of Csk KO mice, as well as MK counts in the bone marrow (BM).2 Recently, biallelic loss-of-function mutations in PTPRJ (g.48131608A>G and g.48158556delG) were described in patients, resulting in a novel form of inherited thrombocytopenia characterized by impaired maturation of MKs and reduced platelet volume, further highlighting the importance of PTPRJ to MK development and function.19
We previously showed that phosphorylation of the C-terminal inhibitory tyrosine residues of SFKs was partially rescued in platelets lacking both Csk and PTPRJ, suggesting that another kinase can compensate for Csk.2 We found that Csk homologous kinase (Chk, also known as megakaryocyte-associated tyrosine kinase, Matk) is upregulated in these platelets and thus might partially fulfill the function of Csk.2 Chk shares ∼50% homology with Csk, with restricted expression in neuronal cells in the brain and hematopoietic cells. Chk was previously detected in platelets by western blotting, but not by mass spectrometry-based approaches, suggesting low levels of expression.20-22 This correlated with the lack of a platelet or hemostatic phenotype in Chk KO mice, drawing into question the functional relevance of Chk in platelets.23 Chk has been implicated in MK progenitor development by using antisense oligonucleotides to reduce protein expression.24 In addition, overexpression of Chk led to a reduction in agonist-induced SFK activity in megakaryocytic cell lines25 and the protein was also implicated in cell spreading26 ; however, in vivo data are lacking.23,27,28
In this study, we have generated and analyzed Chk KO mice alongside Chk;Csk DKO and Chk;Csk;Ptprj triple KO (TKO) mice to determine if there is functional redundancy between Chk and Csk and whether the lack of PTPRJ can rescue MK and platelet phenotypes.
Methods
Mice
Matktm1Sor knockout (Chk KO) mice were from the Jackson Laboratories. Cskfl/fl, Ptprjfl/fl, and Pf4-Cre+ mice were generated, as previously described.29-31 All mice were on a C57BL/6 background. Pf4-Cre+ mice were used as control mice. All procedures were undertaken with United Kingdom Home Office approval in accordance with the Animals (Scientific Procedures) Act of 1986.
Antibodies and reagents
Anti-CD42a antibody was from Abcam; anti-Shp1, anti-Shp1 p-Tyr564, anti-Shp2, and anti-Shp2 p-Tyr580 antibodies were from Cell Signaling Technology; anti-mouse TLT-1 Alexa Fluor-488 antibody was from R&D Systems.32 Fluorescein peanut agglutinin was from Vector Laboratories. Dasatinib was from LC laboratories, PP1 was from Calbiochem, and PRT-060318 was from SynKinase. Other antibodies and reagents were previously described.2
Platelet preparation
Washed platelets were prepared as described.2 Platelet concentration was normalized at 2 × 107/mL for spreading and flow cytometry analysis, and 5 × 108/mL for biochemical analysis.
Saline tail bleeding time assay
Mice were anesthetized using isoflurane and 3 mm of the tail tip was cut off. Immediately, tails were immersed in 0.9% isotonic saline at 37°C. The time to complete arrest of bleeding (no blood flow for 1 minute) was determined. If on/off cycles of bleeding occurred, the sum of bleeding times within a 20-minute period was used. Otherwise, experiments were stopped after 20 minutes.
Immunohistochemistry, platelet functional assays, and biochemistry
Immunohistochemistry, platelet adhesion, stimulation for flow cytometry, and immunoblotting by ProteinSimple Wes are described in detail in supplemental Methods, available on the Blood Web site.
Statistical analysis
Data presented are means ± standard deviation (SD). One-way or 2-way analysis of variance (ANOVA) followed by appropriate post hoc test was used to determine statistical significance (P < .05).
Results
Severe macrothrombocytopenia in Chk;Csk DKO mice
Constitutive Chk KO (CKO) mice were crossed with Cskfl/fl;Pf4-Cre+ mice, in which Csk was conditionally deleted, to generate Chk;Csk DKO mice. DKO mice were further crossed with Ptprjfl/fl mice to generate Chk;Csk;Ptprj TKO mice in which Chk is deleted in every cell, whereas Csk and PTPRJ are deleted in MKs, platelets, and subsets of leukocytes because of leakiness of the Pf4-Cre transgene, as previously described.18 We detected Chk expression in control mouse platelets by quantitative capillary electrophoresis-based immunoassays (ProteinSimple Wes), which was absent in CKO, DKO, and TKO mice (Figure 1A-B). Csk expression was reduced to 6% in DKO and to 1% in TKO platelets compared with control platelets (Figure 1A-C). Surface expression of PTPRJ was reduced to 11% in TKO platelets; however, DKO platelets displayed a mean increase of 166% in PTPRJ surface expression, possibly as a compensatory feedback mechanism (Figure 1D). Platelet counts were normal in CKO mice; however, DKO mice displayed severe thrombocytopenia with a 92% reduction in platelet counts compared with control mice (Figure 1E; supplemental Table 1). This is a further 27% decrease compared with Csk KO mice, previously reported to have a 65% reduction in platelet count.2 Intriguingly, TKO mice had only a 34% reduction in platelet count compared with controls, demonstrating a partial rescue of the DKO phenotype in the absence of PTPRJ (Figure 1E). Mean platelet volume was not significantly different in CKO mice, whereas it was increased 49% in DKO mice and 23% in TKO mice (Figure 1F). The spleen/body weight ratio was also increased by 244% in DKO mice and 115% in TKO mice (Figure 1G), suggesting extramedullary hematopoiesis. These results demonstrate that although Csk can compensate for the loss of Chk in CKO mice, when both kinases are absent, DKO mice develop a more severe thrombocytopenia compared with Csk KO mice, suggesting redundancy in regulating platelet count.
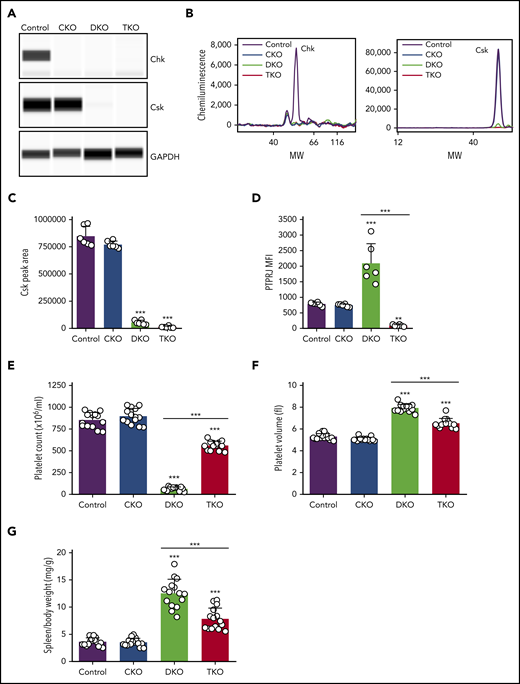
Severe macrothrombocytopenia in DKO mice is partially rescued in TKO mice. (A) Representative blots of capillary-based immunoassays on platelet lysates with the indicated antibodies. (B) Representative data from panel A displayed as electropherograms. (C) Quantification of peak areas of Csk signal from (A-B), n = 6 mice/genotype. (D) Median fluorescence intensity (MFI) measured in αIIb+ cells costained for PTPRJ in blood, n = 6 mice/genotype. (E) Platelet counts, n = 14 mice/genotype. (F) Platelet volumes, n = 14 mice/genotype. (G) Spleen/body weight ratio, n = 15 mice/genotype. Chk−/− (CKO), Chk−/−;Cskfl/fl;Pf4-Cre+ (DKO), Chk−/−;Cskfl/fl;Ptprjfl/fl;Pf4-Cre+ (TKO). *Significant difference compared with control, or significant difference between DKO and TKO. **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD. MW, molecular weight.
Severe macrothrombocytopenia in DKO mice is partially rescued in TKO mice. (A) Representative blots of capillary-based immunoassays on platelet lysates with the indicated antibodies. (B) Representative data from panel A displayed as electropherograms. (C) Quantification of peak areas of Csk signal from (A-B), n = 6 mice/genotype. (D) Median fluorescence intensity (MFI) measured in αIIb+ cells costained for PTPRJ in blood, n = 6 mice/genotype. (E) Platelet counts, n = 14 mice/genotype. (F) Platelet volumes, n = 14 mice/genotype. (G) Spleen/body weight ratio, n = 15 mice/genotype. Chk−/− (CKO), Chk−/−;Cskfl/fl;Pf4-Cre+ (DKO), Chk−/−;Cskfl/fl;Ptprjfl/fl;Pf4-Cre+ (TKO). *Significant difference compared with control, or significant difference between DKO and TKO. **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD. MW, molecular weight.
Aberrant megakaryocyte counts and associated myelofibrosis in DKO and TKO mice
Histological analysis of main sites of thrombopoiesis, namely the BM, spleen, and lung, revealed major differences in MK counts in DKO and TKO mice. We found a 44% increase in MK count per field of view in TKO BM and a 49% increase when counted the total number of MKs per section, but no difference in MK numbers in CKO or DKO BM compared with control mice (Figure 2A, D-E). Notably, MKs in DKO and TKO BM appeared atypical. Furthermore, we found a more than sixfold increase in MK numbers per field of view in the spleen of both DKO and TKO mice (Figure 2B, E). The total number of MKs per spleen section increased by more than 13-fold in DKO and by more than eightfold in TKO mice (Figure 2D). A marginal increase in the number of CD42b+ cells was observed in the lungs of DKO and TKO mice (Figure 2C-D), which may be MKs or MK fragments as a consequence of increased megakaryopoiesis and release from the BM and spleens of these mice. We also observed associated paratrabecular and perivascular reticulin staining in the BM and a marginal increase in reticulin staining in the spleen of DKO and TKO mice (Figure 2A-B), indicative of fibrosis.
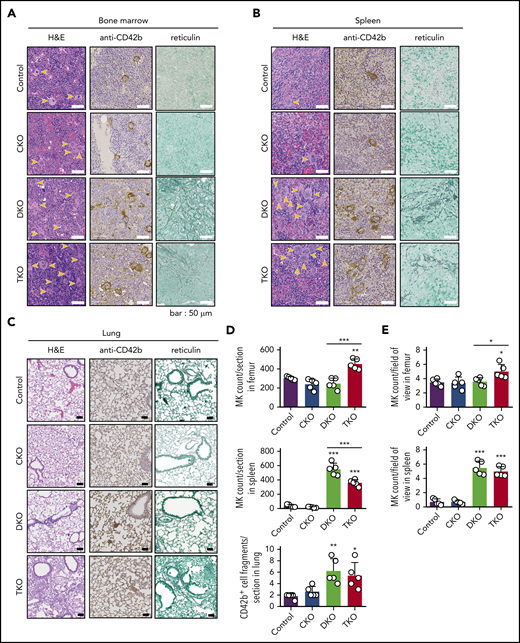
Aberrant megakaryocyte counts and myelofibrosis in DKO and TKO mice. Histological sections of mouse bone marrow (A), spleen (B), and lung (C) of the indicated genotypes stained with hematoxylin and eosin (H&E), the megakaryocytic marker CD42b, or reticulin. MKs are highlighted by yellow arrows. (D) Total number of MKs per section in the indicated organs, n = 5 mice/genotype. (E) MK numbers per field of view in the indicated organs, n = 5 mice/genotype (mean of 15 fields/mouse). Analysis of MK numbers was performed in a double-blinded manner. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
Aberrant megakaryocyte counts and myelofibrosis in DKO and TKO mice. Histological sections of mouse bone marrow (A), spleen (B), and lung (C) of the indicated genotypes stained with hematoxylin and eosin (H&E), the megakaryocytic marker CD42b, or reticulin. MKs are highlighted by yellow arrows. (D) Total number of MKs per section in the indicated organs, n = 5 mice/genotype. (E) MK numbers per field of view in the indicated organs, n = 5 mice/genotype (mean of 15 fields/mouse). Analysis of MK numbers was performed in a double-blinded manner. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
Reduction in the level of GPVI and CLEC-2 in DKO and TKO platelets
To investigate whether changes in platelet count correlate with altered platelet receptors, we quantified surface levels of receptors responsible for platelet production and activation. We found a 21% to 24% increase in αIIbβ3 levels in DKO and TKO platelets, and 21% decrease in α2 levels in DKO platelets (Figure 3A). GPIbα levels were increased by 82% in DKO platelets, which plays a central role in regulating platelet size and count.33 Surface expression of the ITAM-containing GPVI receptor was reduced by 77% and by 97% in DKO and TKO platelets, respectively (Figure 3A). Moreover, surface expression of the hemi-ITAM-containing CLEC-2 receptor was reduced by 43% and 79% in DKO and TKO platelets, respectively. Platelet surface expression of the inhibitory ITIM-containing MPIG6B receptor was reduced by 23% in CKO platelets and increased by 44% in TKO platelets. These data suggest that Chk plays a role in regulating surface expression of ITAM-containing receptors, as previously established for Csk and PTPRJ.2

Reduced expression of (hemi-)ITAM-containing receptors, elevated immature platelet fraction and platelet activation markers in DKO and TKO mice. (A) Median fluorescence intensity (MFI) measured in αIIbβ3+ cells or αIIb+ cells costained for the indicated proteins in blood, n = 6 mice/genotype. (B) Representative image of reticulated platelet population as measured by flow cytometry. αIIb+ platelets in whole blood were gated and the percentage of reticulated platelets was determined in gate P1 (in red), in dot plot diagram forward scatter (FSC) vs RNA dye (Thiazole Orange). (C) Percentage of reticulated platelets as determined in panel B. (D) Representative image of desialylated platelet population. αIIb+ platelets in whole blood were gated and the percentage of desialylated platelets was determined in gate P1 (red), in dot plot diagram FSC vs peanut agglutinin lectin. (E) Percentage of desialylated platelets as determined in panel D. (F) Percentage of P-selectin+αIIb+, TLT-1+αIIb+, fibrinogen+αIIb+, or Annexin V+αIIb+ cells in blood as determined by flow cytometry, n = 6 mice/genotype. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
Reduced expression of (hemi-)ITAM-containing receptors, elevated immature platelet fraction and platelet activation markers in DKO and TKO mice. (A) Median fluorescence intensity (MFI) measured in αIIbβ3+ cells or αIIb+ cells costained for the indicated proteins in blood, n = 6 mice/genotype. (B) Representative image of reticulated platelet population as measured by flow cytometry. αIIb+ platelets in whole blood were gated and the percentage of reticulated platelets was determined in gate P1 (in red), in dot plot diagram forward scatter (FSC) vs RNA dye (Thiazole Orange). (C) Percentage of reticulated platelets as determined in panel B. (D) Representative image of desialylated platelet population. αIIb+ platelets in whole blood were gated and the percentage of desialylated platelets was determined in gate P1 (red), in dot plot diagram FSC vs peanut agglutinin lectin. (E) Percentage of desialylated platelets as determined in panel D. (F) Percentage of P-selectin+αIIb+, TLT-1+αIIb+, fibrinogen+αIIb+, or Annexin V+αIIb+ cells in blood as determined by flow cytometry, n = 6 mice/genotype. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
Increased platelet turnover and preactivation in DKO and TKO mice
To further investigate the cause of thrombocytopenia in DKO and TKO mice, we assessed the fraction of immature reticulated, desialylated and preactivated platelets in whole blood from these mice. All of these parameters were similar in CKO and control mice (Figure 3B-F). However, we found four- and twofold increases in reticulated platelets in DKO and TKO mice, respectively, compared with control mice, suggesting a higher percentage of young platelets in the circulation of these mice (Figure 3B-C). Platelet staining with peanut agglutinin (PNA) lectin, which specifically binds to exposed galactose residues, was used as a marker of desialylation of surface glycans and is associated with old platelets.34 We found a 74% increase in PNA binding to DKO platelets and a 50% increase to TKO platelets compared with controls (Figure 3D-E). We next stained resting platelets for different activation markers. We observed a 52-fold increase in the percentage of P-selectin positive platelets in TKO blood (Figure 3F), suggesting platelet preactivation. Notably, we detected eight- and sevenfold increases in surface TLT-1 levels in DKO and TKO platelets, respectively, compared with control (Figure 3F), providing further evidence of platelet preactivation in these mice. We also observed two- and fivefold increases in fibrinogen binding in DKO and TKO platelets, respectively, compared with platelets from control mice, suggesting the integrin αIIbβ3 was in a high-affinity, active conformation in a proportion of these platelets. We found only a minor increase in the percentage of Annexin V positive platelets in blood from DKO mice (Figure 3F). Increased megakaryocyte counts and immature platelet fraction suggest impaired platelet production in DKO mice, which was partially restored in TKO mice.
Platelet functional defects in DKO mice are rescued in TKO mice
Because of the severe thrombocytopenia in DKO mice, we were only able to test platelet function in a limited set of assays. Platelet α-granule secretion and integrin αIIbβ3 activation were assessed by measuring P-selectin and TLT-1 surface expression and fibrinogen binding, respectively, by flow cytometry. PAR4 peptide-stimulated CKO platelets expressed similar increase in P-selectin and TLT-1 levels and displayed comparable elevation of fibrinogen binding as stimulated control platelets (Figure 4A; supplemental Figure 1). In contrast, DKO platelets exhibited significant impairment in staining for all 3 markers (Figure 4A). Interestingly, PAR4 peptide-stimulated TKO platelets showed a similar increase in P-selectin expression and fibrinogen binding as control platelets, despite higher basal expressions of these markers in whole blood (Figure 3F); however, there was a minor reduction in TLT-1 expression (Figure 4A), presumably as a consequence of activation of inhibitory mechanisms.32

Defective platelet activation and hemostasis in DKO mice is restored in TKO mice. (A) Anti-P-selectin-FITC, anti-triggering receptor expressed on myeloid cells-like transcript 1 (TLT-1)-FITC and fibrinogen-488 binding to washed platelets (2 × 107/mL) following stimulation with or without PAR4 peptide (AYPGKF) (100 μM or 500 μM, 20 minutes, room temperature) was measured by flow cytometry. The fold increase of the MFI relative to the corresponding unstimulated platelets was calculated, n = 5-7 mice/genotype. (B) Representative phalloidin-stained images of resting (basal) and thrombin-stimulated (0.1 U/mL, 5 minutes) platelets spread on fibrinogen-coated cover-slips (100 μg/mL, 45 minutes, 37°C, scale bar: 5 μm). (C) Mean surface area of individual platelets quantified by KNIME software, n = 6 mice/genotype (200 to 450 platelets/condition). (D) Hemostatic response was measured in saline tail bleeding assay by an excision of a 3-mm portion of the tail tip followed by immersion of the tail in 0.9% isotonic saline at 37°C. Plotted is the time to complete arrest of bleeding. Experiments were conducted in a double-blinded manner, n = 16 mice/genotype. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
Defective platelet activation and hemostasis in DKO mice is restored in TKO mice. (A) Anti-P-selectin-FITC, anti-triggering receptor expressed on myeloid cells-like transcript 1 (TLT-1)-FITC and fibrinogen-488 binding to washed platelets (2 × 107/mL) following stimulation with or without PAR4 peptide (AYPGKF) (100 μM or 500 μM, 20 minutes, room temperature) was measured by flow cytometry. The fold increase of the MFI relative to the corresponding unstimulated platelets was calculated, n = 5-7 mice/genotype. (B) Representative phalloidin-stained images of resting (basal) and thrombin-stimulated (0.1 U/mL, 5 minutes) platelets spread on fibrinogen-coated cover-slips (100 μg/mL, 45 minutes, 37°C, scale bar: 5 μm). (C) Mean surface area of individual platelets quantified by KNIME software, n = 6 mice/genotype (200 to 450 platelets/condition). (D) Hemostatic response was measured in saline tail bleeding assay by an excision of a 3-mm portion of the tail tip followed by immersion of the tail in 0.9% isotonic saline at 37°C. Plotted is the time to complete arrest of bleeding. Experiments were conducted in a double-blinded manner, n = 16 mice/genotype. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
We next analyzed platelet spreading on a fibrinogen coated surface as a means of assessing integrin αIIbβ3 function. Resting platelets were either directly plated on fibrinogen or were preactivated with 0.1 U/mL thrombin before plating. CKO platelets spread comparably to control platelets, whereas DKO platelets displayed a 28% increase in spreading (Figure 4B-C). However, in the presence of thrombin, there was no significant difference between control and DKO platelets. Although TKO platelets spread to a similar extent as control platelets, the morphology of thrombin-activated TKO platelets was different compared with control platelets, with reduced lamellipodia and stress fiber formation (Figure 4B).
We next assessed the hemostasis in the various mouse models using the saline tail bleeding assay. We found no significant difference in bleeding time of CKO mice compared with control mice (Figure 4D). In agreement with the 92% reduction in platelet count in DKO mice (Figure 1E), we observed significant bleeding in these mice (Figure 4D). This bleeding phenotype was rescued in TKO mice, which displayed no significant difference in bleeding time compared with control mice (Figure 4D). We conclude that the partially rescued platelet count in combination with the almost normal platelet reactivity in response to thrombin receptor stimulation is sufficient to restore hemostatic function in TKO mice.
Partial rescue of Src and Fyn inhibitory tyrosine phosphorylation in TKO platelets
To study the biochemical consequences of ablating Chk, Csk, and PTPRJ, we assessed tyrosine phosphorylation of SFKs in resting platelets by capillary electrophoresis-based immunoassays. We measured phosphorylation of the activation loop tyrosine residue of SFKs (Src p-Tyr418), an indirect indicator of SFK activity, which was normal in CKO and DKO, and increased 75% in TKO platelets (Figure 5A-B). A possible explanation of normal SFK activity in DKO platelets is increased PTPRJ expression, which can dephosphorylate Src p-Tyr418 and attenuate activity (Figure 1D). We then measured phosphorylation of the C-terminal inhibitory tyrosine residues of Src, Lyn, and Fyn, which binds intramolecularly with their SH2 domain, locking the kinases in an inactive conformation. We observed an 80% to 92% decrease of Src p-Tyr529, Lyn p-Tyr507, and Fyn p-Tyr530 in DKO platelets compared with controls (Figure 5A-B). There was a fourfold increase in Src p-Tyr529 and a twofold increase in Fyn p-Tyr530 in TKO platelets compared with DKO platelets, but no significant difference in Lyn p-Tyr507. These residues were phosphorylated to the same extent in CKO platelets. These findings suggest that another kinase can phosphorylate these inhibitory tyrosine residues in the absence of Chk, Csk, and PTPRJ. In addition, we found that Src, Lyn, and Fyn all exhibited some degree of regulation on the protein level in DKO or TKO platelets. Whereas Src levels were increased by 34% in DKO platelets, Lyn and Fyn levels were reduced by 65% and 34%, respectively, in TKO platelets compared with controls (Figure 5A-B). Moreover, expression of the tyrosine kinase Syk was also found to be reduced in TKO platelets by 37%.
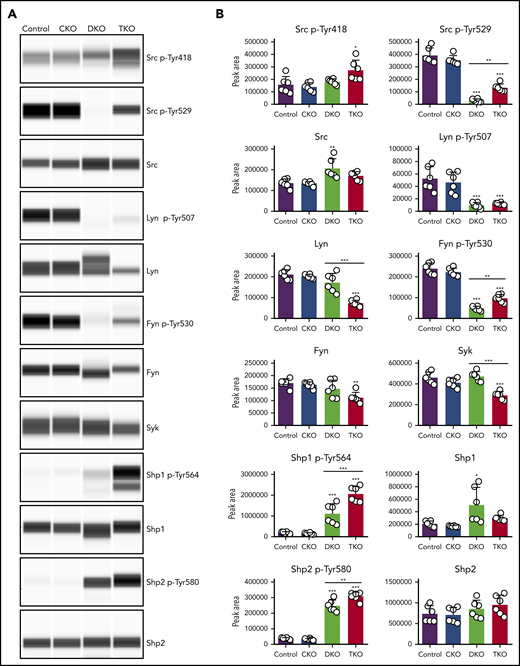
Partial rescue of Src and Fyn inhibitory tyrosine phosphorylation in TKO platelets. (A) Representative blots of capillary-based immunoassays on lysates of resting platelets with the indicated antibodies. (B) Quantification of peak areas, n = 6 mice/genotype. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
Partial rescue of Src and Fyn inhibitory tyrosine phosphorylation in TKO platelets. (A) Representative blots of capillary-based immunoassays on lysates of resting platelets with the indicated antibodies. (B) Quantification of peak areas, n = 6 mice/genotype. *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Sidak test; mean ± SD.
We next assessed the regulation of the cytosolic tyrosine phosphatases Shp1 and Shp2, both of which contain 2 conserved C-terminal tyrosine residues, phosphorylation of which has been implicated in regulating downstream signaling.35-37 We found Shp1 p-Tyr564 to be increased by six- and 10-fold in DKO and TKO platelets, respectively. However, DKO platelets also showed a threefold increase in Shp1 protein levels. Shp2 p-Tyr580 was increased by fivefold in DKO platelets and sevenfold in TKO platelets (Figure 5A-B). These findings demonstrate that SFKs lie upstream of Shp1 and Shp2, acting either directly or indirectly, to activate these phosphatases.
SFKs auto-phosphorylate their C-terminal inhibitory tyrosine residues
To explore which other kinases phosphorylate the C-terminal tyrosine residues of SFKs, we incubated control and TKO platelets with 2 different SFK inhibitors, namely dasatinib and PP1, and the Syk kinase inhibitor PRT-060318.16,38 Dasatinib and PP1 significantly reduced Src p-Tyr418 in control and TKO platelets, whereas PRT-060318 had only a minor effect, as expected. We found that dasatinib reduced Src p-Tyr529, Lyn p-Tyr507, and Fyn p-Tyr530 in TKO platelets by 52% to 62%, and PP1 reduced phosphorylation of these sites by 64% to 73% (Figure 6A-B). These findings suggest that SFKs themselves are able to phosphorylate their C-terminal inhibitory tyrosine residues in the absence of Chk and Csk, thus autoinhibiting their activity (Figure 7). However, PRT-060318 had no significant effect on phosphorylation of these residues in TKO platelets, suggesting Syk plays no role in phosphorylating these residues.
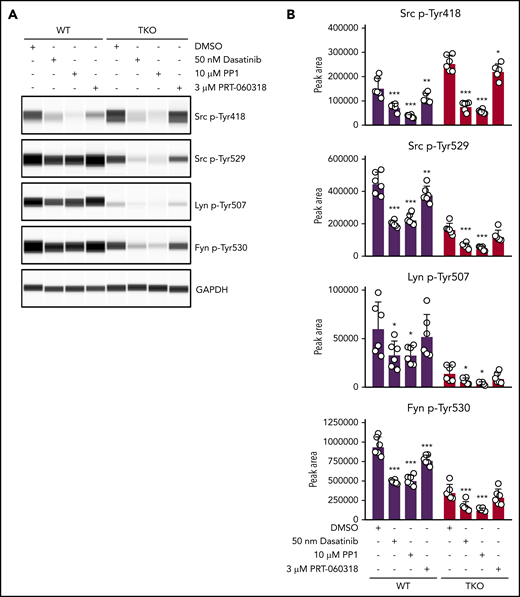
SFKs auto-phosphorylate their C-terminal inhibitory tyrosine residues. (A) Representative blots of capillary-based immunoassays on platelet lysates with the indicated antibodies. Lysates were generated from platelets incubated with DMSO, 50 nM dasatinib, 10 μM PP1, or 3 μM PRT-060318 for 15 minutes, room temperature. (B) Quantification of peak areas, n = 6 mice/genotype. Asterisks refer to significant difference compared with DMSO-treated control samples within genotypes. *P < .05, **P < .01, ***P < .001, 2-way ANOVA with Sidak test; mean ± SD.
SFKs auto-phosphorylate their C-terminal inhibitory tyrosine residues. (A) Representative blots of capillary-based immunoassays on platelet lysates with the indicated antibodies. Lysates were generated from platelets incubated with DMSO, 50 nM dasatinib, 10 μM PP1, or 3 μM PRT-060318 for 15 minutes, room temperature. (B) Quantification of peak areas, n = 6 mice/genotype. Asterisks refer to significant difference compared with DMSO-treated control samples within genotypes. *P < .05, **P < .01, ***P < .001, 2-way ANOVA with Sidak test; mean ± SD.
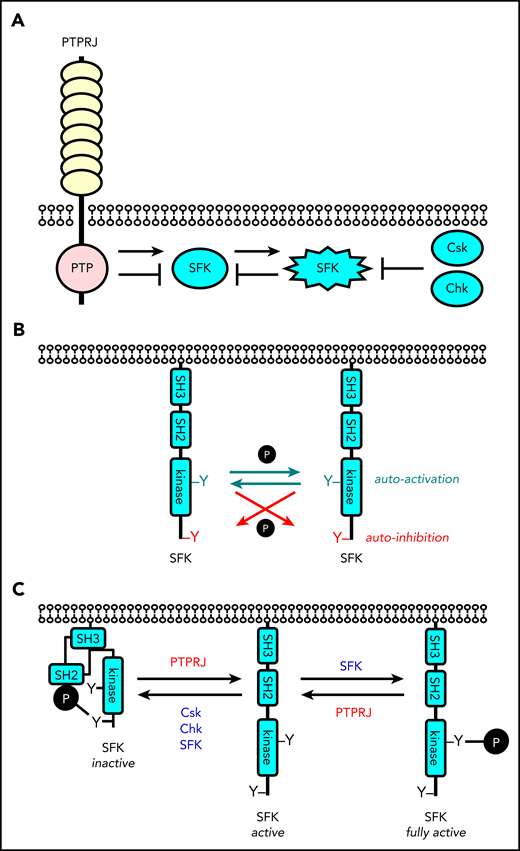
Revised model of regulation of SFKs in the megakaryocyte lineage. (A) SFK activity is tightly regulated by the coordinated action of the tyrosine kinases Csk and Chk, the receptor-type tyrosine phosphatase PTPRJ, and SFKs themselves. Csk and Chk negatively regulate SFK activity, whereas PTPRJ and SFKs are dual positive and negative regulators of SFKs. (B) SFKs autoregulate their catalytic activity through the trans-phosphorylation of conserved tyrosine residues in the activation loop and C-terminal tail. (C) An equilibrium of different states of SFKs is established in resting and activated platelets through the interplay of these tyrosine kinases and phosphatase, as indicated. This is dependent on the concentrations, proximity, and catalytic activities of these enzymes.
Revised model of regulation of SFKs in the megakaryocyte lineage. (A) SFK activity is tightly regulated by the coordinated action of the tyrosine kinases Csk and Chk, the receptor-type tyrosine phosphatase PTPRJ, and SFKs themselves. Csk and Chk negatively regulate SFK activity, whereas PTPRJ and SFKs are dual positive and negative regulators of SFKs. (B) SFKs autoregulate their catalytic activity through the trans-phosphorylation of conserved tyrosine residues in the activation loop and C-terminal tail. (C) An equilibrium of different states of SFKs is established in resting and activated platelets through the interplay of these tyrosine kinases and phosphatase, as indicated. This is dependent on the concentrations, proximity, and catalytic activities of these enzymes.
Discussion
In the present study, we investigated the physiological functions of Chk, Csk, and PTPRJ in platelets. By analyzing mice lacking Chk alone, or in combination with Csk and PTPRJ, we demonstrate that Chk and Csk have redundant functions inhibiting SFKs in the MK lineage (Figure 7), with Csk playing the dominant role, and that PTPRJ plays a central role in both activating and attenuating SFK activity. DKO mice developed severe macrothrombocytopenia with a 92% drop in platelet count, elevated MK numbers, and immature platelet fraction, pointing to disruption of platelet production. Platelet counts in DKO mice were considerably lower than in Csk KO mice, whereas MK numbers in the spleen and immature platelet fraction were double (Table 1).2 These phenotypes suggest that Csk can fully compensate for the absence of Chk in MKs and platelets, whereas Chk can compensate only in part in the absence of Csk. DKO mice revealed that Chk and Csk are essential for platelet production. TKO mice showed partially restored platelet counts, elevated MK numbers in the BM and spleen, and an immature platelet fraction that was between control and DKO mice. These findings together with our previous report demonstrating rescued platelet counts in the Csk;Ptprj DKO mice compared with Csk KO mice highlight that, by controlling the extent of phosphorylation of the C-terminal inhibitory tyrosine residues of SFKs, PTPRJ plays a central role in regulating platelet counts and activation (Figure 7, Table 1).2
Summary of Chk, Csk, and Ptprj knockout mouse phenotypes
| Phenotype | Genotype | |||||
|---|---|---|---|---|---|---|
| Chk KO | Csk KO2 | Ptprj KO2,5 | Chk; Csk DKO | Csk; Ptprj DKO2 | Chk; Csk; Ptprj TKO | |
| Platelet count | — | ↓↓ | — | ↓↓↓ | — | ↓ |
| Reticulated platelets | — | ↑ | — | ↑↑↑ | ↑ | ↑ |
| MK count in bone marrow | — | ↑ | — | — | — | ↑ |
| MK count in spleen | — | ↑↑ | ↑ | ↑↑↑ | ↑↑ | ↑↑↑ |
| Splenomegaly | — | ↑ | — | ↑↑↑ | ↑ | ↑ |
| Myelofibrosis in bone marrow | — | — | — | ↑↑ | — | ↑↑ |
| SFK activity | — | ↑ | ↓↓ | — | ↑↑ | ↑ |
| Platelet receptor expression | ||||||
| GPIbα | — | ↑↑ | — | ↑↑ | ↑ | — |
| GPVI | — | ↓↓ | ↓ | ↓↓ | ↓↓ | ↓↓↓ |
| CLEC-2 | — | ↓ | — | ↓ | ↓↓ | ↓↓ |
| MPIG6B | ↓ | ↑↑ | — | — | ↑↑ | ↑↑ |
| Platelet spreading (fibrinogen) | — | ↑ | ↓ | ↑ | — | — |
| Bleeding | — | ↑ | — | ↑↑↑ | ↑↑ | — |
| Phenotype | Genotype | |||||
|---|---|---|---|---|---|---|
| Chk KO | Csk KO2 | Ptprj KO2,5 | Chk; Csk DKO | Csk; Ptprj DKO2 | Chk; Csk; Ptprj TKO | |
| Platelet count | — | ↓↓ | — | ↓↓↓ | — | ↓ |
| Reticulated platelets | — | ↑ | — | ↑↑↑ | ↑ | ↑ |
| MK count in bone marrow | — | ↑ | — | — | — | ↑ |
| MK count in spleen | — | ↑↑ | ↑ | ↑↑↑ | ↑↑ | ↑↑↑ |
| Splenomegaly | — | ↑ | — | ↑↑↑ | ↑ | ↑ |
| Myelofibrosis in bone marrow | — | — | — | ↑↑ | — | ↑↑ |
| SFK activity | — | ↑ | ↓↓ | — | ↑↑ | ↑ |
| Platelet receptor expression | ||||||
| GPIbα | — | ↑↑ | — | ↑↑ | ↑ | — |
| GPVI | — | ↓↓ | ↓ | ↓↓ | ↓↓ | ↓↓↓ |
| CLEC-2 | — | ↓ | — | ↓ | ↓↓ | ↓↓ |
| MPIG6B | ↓ | ↑↑ | — | — | ↑↑ | ↑↑ |
| Platelet spreading (fibrinogen) | — | ↑ | ↓ | ↑ | — | — |
| Bleeding | — | ↑ | — | ↑↑↑ | ↑↑ | — |
↑ upregulated, ↓ downregulated, — normal.
Biallelic loss-of-function mutations in Ptprj has recently been suggested to cause a novel form of inherited thrombocytopenia in humans.19 Although most of the human findings correlate with results from Ptprj-deficient mice, significant reduction in the platelet count is not observed in constitutive Ptprj loss-of-function transmembrane KO mice or in conditional KO mice generated with either the Pf4-Cre or Gp1ba-Cre transgenes (supplemental Figure 2).2,5,18 Although the cause of these apparent differences is not known, we hypothesize this is primarily because of epigenetic effects and modifier loci in the patients that is not the case in inbred C57Bl/6 mice. Of note, ablation of Ptprj in zebrafish resulted in only a minor drop in thrombocyte counts.19
One of the consequences of increased MK count is the development of myelofibrosis, which was observed in the BM of DKO and TKO mice, but not in Csk KO or Csk;Ptprj DKO mice (Table 1). Primary myelofibrosis is a rare heterogeneous disorder with poor prognosis in which somatic mutations drive clonal expansion of hematopoietic stem cells; and is characterized by BM fibrosis, MK proliferation and atypia, extramedullary hematopoiesis, and splenomegaly. Because MKs play a central role in the development of myelofibrosis,39,40 and PTPRJ plays a central role in regulating MK function,2,5,19 our findings highlight PTPRJ as a potential novel target either alone or in combination with existing therapies in the treatment of myeloproliferative neoplasms and myelofibrosis.
Phosphorylation of C-terminal inhibitory tyrosine residues was barely detectable in DKO platelets, which increased significantly in Src and Fyn in TKO platelets, demonstrating phosphorylation in the absence of the only 2 known kinases that are able to phosphorylate these sites.41 It also illustrates that PTPRJ must constitutively dephosphorylate these residues, even in resting platelets, maintaining a pool of active SFKs in platelets, facilitating rapid activation following vascular injury. We observed that the levels of total Lyn were reduced by 65% in TKO platelets, which may explain the lack of increase in phosphorylation of Tyr507 in its C-terminal tail. The mechanism for the reduction of Lyn expression is not known, but likely involves degradation by the E3 ubiquitin ligase c-Cbl, as previously described in immune cells.42 Inhibition of an analog-sensitive variant of Csk induced robust c-Cbl phosphorylation and rapid LynA degradation in macrophages.42 In spite of the lack of C-terminal inhibitory phosphorylation of SFKs in DKO platelets, we did not observe a significant increase in their activation loop phosphorylation. The reason for this could be the significant increase in PTPRJ levels in these platelets, which is able to dephosphorylate the activation loop tyrosine of SFKs, in addition to the C-terminal inhibitory tyrosine residues.2,6 The increase in Shp1 p-Tyr564 and Shp2 p-Tyr580 suggests elevated inhibitory signaling in DKO and TKO platelets. Shp1 phosphorylation has previously been shown to be increased in macrophages expressing a hyperactive variant of Lyn and to be diminished in Lyn KO cells.43 Specifically, Lyn has been demonstrated to be the predominant kinase phosphorylating Shp1 p-Tyr564 site.44 Src was also shown to directly phosphorylate Shp1.35 In addition, thrombin-induced Shp1 phosphorylation was absent in the presence of SFK inhibitors in platelets.45 Similarly, Shp2 p-Tyr580 may also be a direct substrate of SFKs. These studies suggest that SFK activity was indeed elevated in DKO platelets, despite normal phosphorylation of the SFK activation loop tyrosine residue.
Inhibitor studies using TKO platelets indicate that SFKs are capable of auto-phosphorylating their C-terminal tails and presumably autoinhibiting their activity (Figure 7B). In addition to SFKs, both PP1 and dasatinib inhibit Csk at higher concentrations, whereas the latter also blocks the activity of the tyrosine kinases Btk, Tec, and Abl.46-48 Although we cannot exclude the possibility that inhibition of Btk, Tec, or Abl may contribute to the reduction of C-terminal inhibitory site phosphorylation of SFKs in the dasatinib-treated samples, the activity of these kinases should not be affected in the PP1-treated samples. Interestingly, we also observed a significant decrease in the inhibitory site phosphorylation of Src, Lyn, and Fyn in control platelets incubated with SFK inhibitors, which may be the consequence of the combined decrease in SFK and Csk activities. We hypothesize that the autoinhibitory function is particularly important as a means of preventing excessive and prolonged SFK activation in both unstimulated and ligand-mediated receptor clustering conditions.
Because SFKs are ubiquitously expressed, auto-phosphorylation of their C-terminal inhibitory site has previously not appreciated broad implications for several cell types.41,49 Indeed, deletion of Chk alone had no detectable effect on phosphorylation of the C-terminal inhibitory tyrosine residues of SFKs in platelets, demonstrating that Csk and autoinhibition is sufficient to prevent aberrant SFK activation. Residual SFK-mediated auto-phosphorylation of the inhibitory tyrosine residues in the absence of Chk, Csk, and PTPRJ correlated with normal bleeding in these mice, providing evidence of the physiological relevance of this mechanism. The kinetics of phosphorylation of these 2 regulatory tyrosine residues requires further investigation to better understand how SFKs autoregulate their activity. Previous work from T cells has revealed a complex interplay between membrane localization and phosphorylation of these residues in T-cell receptor signaling, requiring further investigation in platelets.50
Both DKO and TKO platelets showed signs of preactivation in the absence of stimuli, highlighted by increased TLT-1 expression and fibrinogen binding. Interestingly, in response to PAR4 stimulation, TKO platelets were able to secrete their granules and bind fibrinogen similarly to control platelets, possibly due to the remaining inhibitory phosphorylation of SFKs. This may also be the result of elevated Shp1 and Shp2 inhibitory activity, via the ITIM-containing receptor MPIG6B.2,51-53 In contrast, DKO platelets lacking phosphorylation of the C-terminal inhibitory tyrosine residues of SFKs were unable to respond to thrombin receptor stimulation. As a consequence of reduced platelet count and reactivity, DKO mice exhibited significantly increased tail bleeding, whereas TKO mice responded similarly to control mice in this assay. Increased PNA staining of platelets from DKO and TKO mice, indicative of increased desialylation of surface glycans, suggested aberrant platelet turnover, with a population of platelets remaining in the circulation longer than normal. Intriguingly, we observed significant downregulation of (hemi-)ITAM containing receptors in TKO platelets with only 3% of GPVI levels and 21% of CLEC-2 levels. These findings reinforce the key role of SFKs in regulating surface GPVI and CLEC-2 levels previously observed in Csk KO and Csk;Ptprj DKO platelets (Table 1),2 which might be explained by potentially increased activation of c-Cbl.42 Alternatively, decreased GPVI and CLEC-2 levels might be a consequence of platelet preactivation. Of note, concomitant deletion of these (hemi-)ITAM-containing receptors in mice has previously been shown to result in a profound bleeding diathesis,54 which is likely counteracted by elevated SFK activity in TKO platelets.
In summary, we demonstrated a critical role for Chk, Csk, and PTPRJ in platelet homeostasis. Although Chk was previously implicated in MK maturation, in vivo evidence was lacking.23-28 In our study, we demonstrate no megakaryocyte or platelet functional defects in CKO mice. However, DKO mice developed severe macrothrombocytopenia, elevated MK counts, and immature platelet fraction and myelofibrosis, pointing to an important role of Chk and Csk in regulation of platelet counts. Importantly, additional deletion of PTPRJ from MKs and platelets partially corrected platelet counts and immature platelet fraction. Because tyrosine phosphatases are increasingly considered druggable, targeting PTPRJ might serve as an alternative strategy to regulate platelet counts in instances of high SFK activity.55 We have also identified a novel inhibitory mechanism, involving auto-phosphorylation of the C-terminal inhibitory tyrosine residues of SFKs, which likely plays an important role in regulating these kinases in other cell types, including immune and cancer cells. In platelets, the partially rescued inhibitory phosphorylation and elevated platelet counts preserved the ability of platelets to respond to stimuli and restored hemostasis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge Arthur Weiss (University of California, San Francisco, CA) for providing Ptprjfl/fl mice,30 Alexander Tarakhovsky (The Rockefeller University, New York, NY) for providing Cskfl/fl mice,29 Jeremy Pike (University of Birmingham, Birmingham, United Kingdom) for providing the KINME analysis software, Gordon Stamp, Mahrokh Nohadani, and Rieda Ahmad (Advance Histopathology Laboratory Ltd, London, United Kingdom) for performing histology analysis and all members of the Birmingham Biomedical Sciences Unit for maintenance of mouse colonies. The authors thank Oreste Acuto (University of Oxford, Oxford, United Kingdom) for intellectual contributions.
This work was supported by British Heart Foundation (BHF) Senior Basic Science Research Fellowship FS/13/1/29894 and BHF Programme Grant RG/15/13/31673 (Y.A.S.) and by a Research Development Fund provided by the University of Birmingham (Z.N.). Z.N. and J.M. are BHF postdoctoral research associates (RG/15/13/31673) and A.M. is a BHF Intermediate Basic Science Research Fellow (FS/15/58/31784).
Authorship
Contribution: Y.A.S. provided conceptualization; Z.N., J.M., and Y.A.S. provided methodology; Z.N., J.M., V.-S.I., and Y.A.S. provided the investigation; Y.A.S. provided resources; A.M. and Y.A.S. provided mouse models and reagents; Z.N. and Y.A.S. wrote the original draft; Z.N., J.M., V.-S.I., A.M., and Y.A.S. performed review and editing; Y.A.S. provided supervision; and Z.N. and Y.A.S. provided funding acquisition.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yotis A. Senis, Université de Strasbourg, Institut National de la Santé et de la Recherche Médicale, Unité Mixte de Recherche–S1255, Etablissement Français du Sang Grand-Est, 10 Rue Spielmann, 67065 Strasbourg, France; e-mail: yotis.senis@efs.sante.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal