

TO THE EDITOR:
Acquired mutations leading to aberrant activation of JAK/STAT signaling are common in myeloproliferative neoplasms (MPNs).1 Ruxolitinib is a JAK1/2 tyrosine kinase inhibitor used to treat patients with certain MPNs.2-6 The US Food and Drug Administration originally approved ruxolitinib for the treatment of advanced myelofibrosis (MF) because of its efficacy reducing symptoms and splenomegaly in this morbid disease.4-6 Weight gain among cachectic patients treated with ruxolitinib was quickly identified and reported among measures of improved quality of life.4-8 Although the original rationale was to disrupt pathogenic signaling via the mutated JAK2(V617F), ruxolitinib proved efficacious in patients with or without this mutation. Ruxolitinib use has since become more widespread. Now with US Food and Drug Administration approval for polycythemia vera2 and graft-versus-host disease9,10 and with an ever-expanding pool of indications being sought,11,12 many patients and clinicians recognize that weight gain and hyperlipidemia are a common consequence of ruxolitinib use.4-8 Yet the reason for these metabolic effects is currently, entirely unknown. This led us to explore the mechanism for ruxolitinib activity on body weight in patients with MPNs.
The study cohort was comprised of all patients treated at Weill Cornell Medicine receiving ruxolitinib for a World Health Organization MPN diagnosis at the time of data closure (November 2019). Subjects were identified by unbiased electronic query using standardized tools and methods (supplemental Materials and methods, available on the Blood Web site). All patients with confirmed MPN diagnosis during ruxolitinib treatment were included. Patients taking ruxolitinib for graft-versus-host disease after hematopoietic stem cell transplant were removed from the cohort, as were patients receiving ruxolitinib only after transformation to an acute leukemia. No data were analyzed before prospective identification of the study cohort. The final cohort comprised 179 patients.
Demographics, vital signs, laboratory values, and concomitant medications were collected by electronic queries at baseline and during treatment. Patients received ruxolitinib for a median of 120 weeks (130-160 weeks, 95% confidence interval [CI]), during which 124 (69%) gained weight (Figure 1A-B). The median weight gain among all patients was 6.7% (95% CI, 3.6%-7.0%) of starting weight with the average weight gain being 12% (95% CI, 10%-13%) among patients gaining any weight. Thus, weight gain was common and substantial.
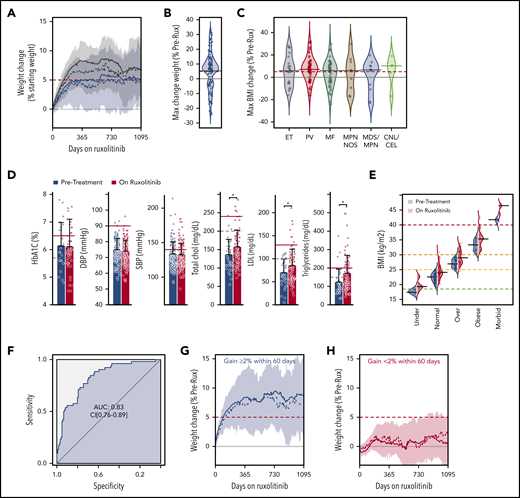
Weight gain among patients with MPN taking ruxolitinib is independent of diagnosis or BMI group. (A) Weight change is shown for patients taking ruxolitinib; presented as percent of the starting weight. The average (solid line), median (dashed line), and interquartile range (shaded) weight change is shown for all 179 patients (blue) and for the 124 patients who gained weight after ruxolitinib was started (gray). (B) Bean plot shows the maximum percent change in weight for all patients after starting ruxolitinib with the group median (blue line) and 5% change (red dashed line) indicated. (C) Bean plots showing the maximum percent BMI change for patients with the indicated MPN diagnoses. Indicator lines are shown as for panel B. (D) Metabolic parameters and blood pressure is shown for patients before (blue) and after (red) starting ruxolitinib. The upper limit of normal (dashed line) and clinically significant elevation (red line) are indicated (statistical significance of key differences indicated, Student t test *P < .05). (E) The BMI of patients before (blue) and after (red) starting ruxolitinib is shown for all patients grouped by their World Health Organization BMI category at the start of therapy. (F) Receiver operating characteristic analysis for weight change within the first 60 days of ruxolitinib as a predictor of gaining more than 10% of starting weight, becoming overweight or increasing BMI category during course of treatment. The change in weight during course of treatment is shown for patients gaining (G) ≥2% or (H) <2% of their starting weight within 60 days of initiating ruxolitinib. AUC, area under the curve; CEL, chronic eosinophilic leukemia; chol, cholesterol; CNL, chronic neutrophilic leukemia; DBP, diastolic blood pressure; ET, essential thrombocythemia; LDL, low-density lipoprotein; MDS/MPN, myelodysplastic/myeloproliferative neoplasm; MF, myelofibrosis; NOS, unclassifiable; PV, polycythemia vera; SBP, systolic blood pressure.
Weight gain among patients with MPN taking ruxolitinib is independent of diagnosis or BMI group. (A) Weight change is shown for patients taking ruxolitinib; presented as percent of the starting weight. The average (solid line), median (dashed line), and interquartile range (shaded) weight change is shown for all 179 patients (blue) and for the 124 patients who gained weight after ruxolitinib was started (gray). (B) Bean plot shows the maximum percent change in weight for all patients after starting ruxolitinib with the group median (blue line) and 5% change (red dashed line) indicated. (C) Bean plots showing the maximum percent BMI change for patients with the indicated MPN diagnoses. Indicator lines are shown as for panel B. (D) Metabolic parameters and blood pressure is shown for patients before (blue) and after (red) starting ruxolitinib. The upper limit of normal (dashed line) and clinically significant elevation (red line) are indicated (statistical significance of key differences indicated, Student t test *P < .05). (E) The BMI of patients before (blue) and after (red) starting ruxolitinib is shown for all patients grouped by their World Health Organization BMI category at the start of therapy. (F) Receiver operating characteristic analysis for weight change within the first 60 days of ruxolitinib as a predictor of gaining more than 10% of starting weight, becoming overweight or increasing BMI category during course of treatment. The change in weight during course of treatment is shown for patients gaining (G) ≥2% or (H) <2% of their starting weight within 60 days of initiating ruxolitinib. AUC, area under the curve; CEL, chronic eosinophilic leukemia; chol, cholesterol; CNL, chronic neutrophilic leukemia; DBP, diastolic blood pressure; ET, essential thrombocythemia; LDL, low-density lipoprotein; MDS/MPN, myelodysplastic/myeloproliferative neoplasm; MF, myelofibrosis; NOS, unclassifiable; PV, polycythemia vera; SBP, systolic blood pressure.
Although anorexia and early satiety resulting from aberrant cytokines and massive splenomegaly are commonly implicated in the weight loss observed in patients with advanced myelofibrosis, cachexia is far less commonly found in patients with MPNs other than MF. Nonetheless, we found that patients with both indolent and more morbid MPNs gained considerable weight (Figure 1C), suggesting that this weight gain did not solely result from correction of MPN-linked symptoms.
As reported by Mesa et al,7,13 total and low-density lipoprotein cholesterol were higher in patients treated with ruxolitinib compared with pretreatment measures from the same group of patients (Figure 1D). We found triglycerides also increased while on ruxolitinib. Although the increase in cholesterol was relatively minor for most patients, a greater proportion of patients had hyperlipidemia when taking ruxolitinib. Median random serum glucose was slightly higher in patients taking ruxolitinib (102 vs 106 mg/dL, P < .01) but we did not find ruxolitinib use associated with changes in cardiovascular risk factors such as diabetes (hemoglobin A1C) or hypertension (Figure 1D).
Whereas only 7 patients (4%) were underweight as determined by body mass index (BMI), 75 patients (42%) were already overweight when beginning treatment with ruxolitinib. Patients gained significant weight regardless of their pretreatment BMI category (Figure 1E) and 64 (36%) patients increased their BMI category during ruxolitinib therapy (Figure 1E). Thus, normalization of suppressed appetite was an unlikely explanation for the weight gain observed.
Appetite is elaborately regulated, and leptin signaling is centrally involved in a complex homeostatic mechanism controlling hunger, satiety, metabolism, and body weight.14-17 Leptin serves as a feedback signal reporting nutritional state to the brain. After feeding, leptin levels rise and this reduces appetite and feeding (supplemental Figure 1). Disturbances of leptin signaling are associated with obesity, hyperlipidemia, and elevated plasma glucose. Leptin exerts many of its effects through leptin receptors (LEPR) expressed on specialized neurons within the arcuate nucleus of the hypothalamus (ARC).18-23 Because the effects of hypothalamic leptin receptor signaling is largely mediated by JAK2/STAT3,24,25 we wondered whether disruption of this pathway by ruxolitinib could be responsible for the weight gain observed in patients.
We therefore developed a mouse model. Eight-week-old male C57BL6/J mice (Jackson Laboratories) were randomized into 4 groups: (1) fasted (Fasted); (2) fasted+leptin (Fasted+Lep); (3) fed (Fed); and 4) fed+ruxolitinib (Fed+Rux). Mice were fasted overnight as a negative control for leptin signaling. Recombinant leptin (5 mg/kg, Peprotech) was administered intraperitoneally to fasted mice 1 hour before harvest as a postive control. Fed mice were provided access to their usual chow and were administered either ruxolitinib (60 mg/kg) or phosphate-buffered saline vehicle control the evening before harvest. The ruxolitinib dose (60 mg/kg) used is roughly equivalent to the human 20 mg twice-daily dose and has been previously shown sufficient for central nervous system penetration.26,27
All mice were euthanized and perfused with 4% paraformaldehyde and their brains were harvested, dehydrated with 30% sucrose, and cryopreserved (OCT, Tissue-Tek). Position-matched 20 μm floating coronal sections were washed, pretreated, and then blocked in normal serum. LEPR signaling in brain was reported by immunofluorescence staining for STAT3 phosphorylation (pSTAT3) using rabbit monoclonal (Cell Signaling cat. #9145S) primary and Cy3 labeled anti-rabbit secondary antibodies (Jackson ImmunoResearch, cat. #711-165-152). Immunofluorescent images were acquired (Zeiss, CSU-X1 Confocal Spinning Disc microscope) using identical conditions for all sections and pSTAT3 signal intensity within the ARC (Figure 2A-B) was quantified using custom scripts and available R packages (EBImage) (supplemental Figure 2). Tissue processing, staining, imaging, and analysis was done blinded to investigators.
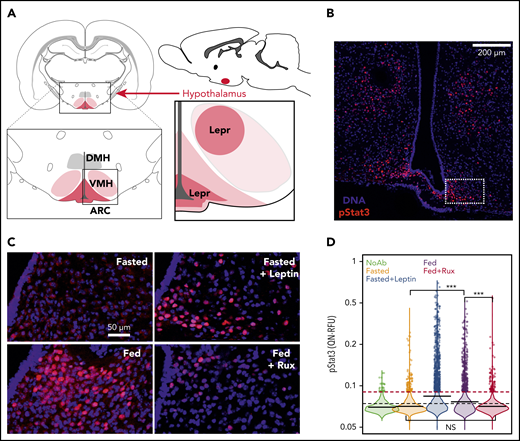
Ruxolitinib blocks postprandial leptin signaling in the hypothalamus. (A) Schematic localizing the LEPR-expressing neurons within the ventromedial hypothalamus (VMH) and arcuate nucleus (ARC). The lower right subpanel represents the region shown in Panel B. (B) A representative image is shown of an 8-week-old male C57BL6/J fasted mouse treated with Leptin (scale bar, 200 μm). The inset box (dashed) shows the region analyzed in panels C-D. (C) Representative images of ARC from mice that were Fasted, Fasted+Lep, Fed, and Fed+Rux (scale bar, 50 μm). (D) The quantile normalized segmented nuclear pSTAT3 signal intensity is shown for replicate animals treated as described (n = 3-5 mice per group, statistical significance of key differences indicated, t test ***P < 10−15). All experiments were performed in accordance with institutional guidelines and were approved by the International Animal Care and Use Committee and in compliance with the Research Animal Resource Center in the Weill Cornell vivarium. DMH, dorsomedial hypothalamus; NS, not significant.
Ruxolitinib blocks postprandial leptin signaling in the hypothalamus. (A) Schematic localizing the LEPR-expressing neurons within the ventromedial hypothalamus (VMH) and arcuate nucleus (ARC). The lower right subpanel represents the region shown in Panel B. (B) A representative image is shown of an 8-week-old male C57BL6/J fasted mouse treated with Leptin (scale bar, 200 μm). The inset box (dashed) shows the region analyzed in panels C-D. (C) Representative images of ARC from mice that were Fasted, Fasted+Lep, Fed, and Fed+Rux (scale bar, 50 μm). (D) The quantile normalized segmented nuclear pSTAT3 signal intensity is shown for replicate animals treated as described (n = 3-5 mice per group, statistical significance of key differences indicated, t test ***P < 10−15). All experiments were performed in accordance with institutional guidelines and were approved by the International Animal Care and Use Committee and in compliance with the Research Animal Resource Center in the Weill Cornell vivarium. DMH, dorsomedial hypothalamus; NS, not significant.
Fasted mice had low pSTAT3 that could be induced by exogenous leptin to a level seen in Fed mice (Figure 2C-D). However, the robust pSTAT3 staining observed in Fed mice could be completely blocked by ruxolitinib (Figure 2D). Indeed, pSTAT3 signaling in the Fed+Rux cohort was indistinguishable from Fasted mice. Ruxolitinib could also block signaling in fasted mice treated with exogenous leptin suggesting that the inhibition of feeding-induced pSTAT3 signaling was largely due to blockade of leptin’s effects (supplemental Figure 3). These results offer a mechanistic explanation for the weight gain and increased cholesterol observed in patients taking ruxolitinib.7,22
Leptin is secreted by adipocytes in amounts directly correlated with the quantity of stored fat.28,29 Thus, leptin levels typically rise when body fat increases. Accordingly, overweight people tend to have high leptin levels.30-32 Patients treated on the Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment (COMFORT) I and II studies gained an average of ∼4 kg and were almost 6 kg heavier than those on the control arm at the time of analysis.4,5,7 Thus, weight gain offers a plausible explanation for the increased plasma leptin levels reported in these foundational studies.
Although many patients with MPNs benefit greatly from ruxolitinib, some also increase cardiovascular risk by gaining substantial weight and increasing serum cholesterol. We searched for a simple, clinically useful predictive model to identify patients with the greatest risk and found those gaining ≥2% of their pretreatment weight within 60 days of starting ruxolitinib treatment were more likely to continue gaining weight, increase BMI category and/or become overweight (Figure 1F-G).
In conclusion, on-target activity of ruxolitinib can abrogate postprandial leptin signaling and in so doing, cause hyperphagia and contribute to the weight gain experienced by most patients. Thus, physicians beginning ruxolitinib treatment should inform their patients of this drug effect and help mitigate adverse outcomes by offering appropriate dietary counseling and lifestyle management recommendations to those with early weight gain.
The online version of this article contains a data supplement.
Acknowledgment
This work was funded by the Johns Family Foundation of the Cancer Research & Treatment Fund, New York, NY.
Authorship
Contribution: J.M.S. designed research; N.M., S.K., P.M., R.T.S., E.R., and J.M.S. performed research and wrote the paper; and P.K., S.K., and J.M.S. analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Joseph M. Scandura, Weill-Cornell Medicine, 1300 York Ave, C610D, Box 113, New York, NY 10065; e-mail: jms2003@med.cornell.edu.
