

Key Points
Multiple engineering steps led to efficient recycling, antigen disposal, and infrequent low-volume subcutaneous self-administration of crovalimab.
PNH patients, treatment naive or switching from SoC, were stably controlled on up to every 4-week subcutaneous self-administered injections of crovalimab.

Abstract
Complement C5 inhibition is the standard of care (SoC) for patients with paroxysmal nocturnal hemoglobinuria (PNH) with significant clinical symptoms. Constant and complete suppression of the terminal complement pathway and the high serum concentration of C5 pose challenges to drug development that result in IV-only treatment options. Crovalimab, a sequential monoclonal antibody recycling technology antibody was engineered for extended self-administered subcutaneous dosing of small volumes in diseases amenable for C5 inhibition. A 3-part open-label adaptive phase 1/2 trial was conducted to assess safety, pharmacokinetics, pharmacodynamics, and exploratory efficacy in healthy volunteers (part 1), as well as in complement blockade–naive (part 2) and C5 inhibitor–treated (part 3) PNH patients. Twenty-nine patients were included in part 2 (n = 10) and part 3 (n = 19). Crovalimab concentrations exceeded the prespecified 100-µg/mL level and resulted in complete and sustained terminal complement pathway inhibition in treatment-naive and C5 inhibitor–pretreated PNH patients. Hemolytic activity and free C5 levels were suppressed below clinically relevant thresholds (liposome assay <10 U/mL and <50 ng/mL, respectively). Safety was consistent with the known profile of C5 inhibition. As expected, formation of drug-target-drug complexes was observed in all 19 patients switching to crovalimab, manifesting as transient mild or moderate vasculitic skin reactions in 2 of 19 participants. Both events resolved under continued treatment with crovalimab. Subcutaneous crovalimab (680 mg; 4 mL), administered once every 4 weeks, provides complete and sustained terminal complement pathway inhibition in patients with PNH, warranting further clinical development (ClinicalTrials.gov identifier, NCT03157635).
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a life-threatening syndrome with sudden onset of hematuria, anemia, and thrombosis.
Eculizumab1 and ravulizumab2,3 (recently approved in the United States, European Union, and Japan) are used to treat PNH; they reduce intravascular hemolysis and improve quality of life (QoL) and, likely, survival.4,5 However, a prospective cohort study reported that IV eculizumab, at the label dose of 900 mg every 2 weeks, was not effective in all patients, delivering detectable hemolytic activity (CH50 > 10%) in 49% of patients6 and increasing the likelihood of pharmacodynamic (PD) breakthrough symptoms due to low exposure. This results in a significant fraction (20%-40%) of patients being treated with eculizumab at higher-than-label dose.6 In addition, no published data exist on patients treated with 1200 mg of eculizumab every 2 weeks switching to ravulizumab, because these patients were excluded from the pivotal trials.2,3 For these patients, only IV treatment options are available at this time.2 Furthermore, a single missense C5 heterozygous mutation (c.2654G→A, single nucleotide polymorphism [SNP]) in ∼3% of the Japanese population is associated with a lack of response to eculizumab7 and, likely, to ravulizumab as well, because both bind to the same epitope. Another limitation of currently available treatments is the need for time- and resource-consuming clinic visits for patients or home nurse visits for lifelong IV administration, which in some cases is a barrier to starting and/or adhering to treatment.
Crovalimab (RO7112689 or SKY59; Chugai Pharmaceutical) is a novel anti-C5 sequential monoclonal antibody recycling technology (SMART) antibody that combines isoelectric point, neonatal Fc receptor, and pH-dependent affinity engineering. This results in efficient C5 binding, enhanced uptake of C5-bound crovalimab by endothelial cells, disposal of C5 in the endosome, and efficient neonatal Fc receptor–mediated recycling of crovalimab (supplemental Figure 1, available on the Blood Web site). Furthermore, crovalimab is highly soluble, allowing for small injection volumes. Crovalimab binds to the C5 β-chain and prevents cleavage of the wild-type and SNP C5 by the C5 convertase. In addition, crovalimab uniquely inhibits C5b6 deposition on membranes,8,9 further limiting membrane attack complex–mediated tissue damage. SMART has led to limited C5 accumulation and increased C5 binding capacity in nonhuman primates.8 These data indicated the possibility of crovalimab having similar or better efficacy than standard of care (SoC) with a smaller subcutaneously administrable amount of drug. Drug-target-drug complexes (DTDCs) are expected to develop if patients are exposed to crovalimab and eculizumab simultaneously (supplemental Figure 2), during a switch period from 1 drug to the other due to the differential epitope recognition of C5 by the antibodies. Being immune complexes in the widest sense, DTDCs could potentially impact safety or efficacy during the switch phase.
COMPOSER (NCT03157635) is a phase 1/2 3-part adaptive design clinical trial to assess the safety, tolerability, PD, and pharmacokinetics (PK) of crovalimab in healthy volunteers (HVs) and patients with PNH, as well as the efficacy, immunogenicity, and patient-reported outcomes of crovalimab in patients with PNH. The first-in-human results of dose-ascending part 110 showed that crovalimab was safe and well tolerated, and the PK and PD profiles supported further investigation. Results from part 2 (complement blockade–naive patients) and part 3 (C5 inhibitor–treated patients) of COMPOSER are reported.
Material and methods
Trial design and oversight
COMPOSER was conducted in compliance with good clinical practice, the principles of the Declaration of Helsinki, according to a written protocol approved by the institutional review board for each participating center. The sponsors, F. Hoffmann-La Roche Ltd. and Chugai Pharmaceutical, developed the protocol in cooperation with the investigators. Full details of the trial design and analyses are provided in supplemental Details.
COMPOSER is a 3-part open-label adaptive phase 1/2 trial that was conducted to assess safety, PK, PD, and exploratory efficacy in HVs (part 1), as well as in complement blockade–naive PNH patients (part 2) and C5 inhibitor–treated PNH patients (part 3).
Participants
Patients aged 18 to 75 years with a PNH clone size >10% by red blood cells (RBCs) or granulocytes within the previous 3 months were eligible for the study. Patients enrolled in part 2 were treatment naive and included patients with no or time-limited access to eculizumab because of reimbursement limitations. Other patients had not wanted to engage in the lifelong nature of necessary biweekly travel to transfusion centers for IV treatment. Patients enrolled in part 3 had been treated with eculizumab previously for ≥3 months. All patients had been vaccinated against Neisseria meningitidis and provided written informed consent.
Interventions
In part 2, IV intrapatient dose escalation of crovalimab (375 mg on day 1, 500 mg on day 8, and 1000 mg on day 22) was followed by weekly subcutaneous crovalimab (170 mg; 1 mL), starting on day 29. In part 3, patients were administered an IV loading dose (1000 mg) of crovalimab 2 weeks after the last dose of eculizumab and then randomized in a 1:1:1 ratio to 1 of 3 subcutaneous crovalimab regimens for 20 weeks: 680 mg (2 × 2 mL) every 4 weeks, 340 mg (2 mL) every 2 weeks, or 170 mg (1 mL) weekly, starting on day 8. Because 2 mild-moderate adverse events (AEs) occurred, which were possibly related to crovalimab, in the first 2 patients in the 680-mg arm, the dose schedule in this arm was adapted to include 8 weekly injections (170 mg) before switching to the 680-mg every-4-week schedule. Also, inclusion criteria for all patients were adapted to exclude patients with an elevated risk for immune complex–mediated disease. A 3-year extension study (open-label extension [OLE]) was available for patients after completing 20 weeks of part 2 or part 3.
Assessments and outcomes
Safety was monitored by physical examination, vital signs, electrocardiogram, laboratory tests, and recording of AEs. Lactate dehydrogenase (LDH) and hemoglobin were assessed locally and standardized to Système International units.11 Patient-reported outcome data on health-related QoL (HRQoL) were collected to inform treatment efficacy. Serum crovalimab, total C5 (tC5) concentrations, and anti-drug antibodies (ADAs) were measured using an enzyme-linked immunosorbent assay (ELISA). The amount of C5 not bound to crovalimab-free C5 (fC5) was analyzed using a fluorophore-based immunoassay.
Complement activity was assessed by a liposome immunoassay (LIA; Wako Autokit CH 50; FUJIFILM Wako Chemicals Europe, Neuss, Germany).12 Complete complement inhibition was defined as CH50 <10 U/mL.
The size and temporal distribution of DTDCs in serum samples were analyzed by a combination of size-exclusion chromatography with a crovalimab-specific ELISA.13
HRQoL was assessed using the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) and European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30; supplemental Details) scales.
Efficacy outcome measures
Efficacy measures included change in LDH, proportion of patients with stabilized hemoglobin, change in fatigue, number of RBC units transfused per patient, and proportion of patients with complement suppression.
Breakthrough hemolysis (BTH) was not predefined in the protocol, and BTH events were reported by the investigators as AEs. The criteria reported by Kulasekararaj et al2 were used for post hoc analysis.
Objectives and statistical analysis
Sample sizes in parts 2 and 3 were chosen for initial safety analysis and robust PK-PD modeling. All analyses were descriptive. The prespecified primary objectives were safety, tolerability, and PD effect on complement activity. Secondary objectives were change in LDH, proportion of patients with stabilized hemoglobin, and proportion of transfusion-free patients. PK data from parts 2 and 3 were pooled and analyzed using a nonlinear mixed-effects approach. Exploratory HRQoL end points were change from baseline in the FACIT-Fatigue and EORTC QLQ-C30 scales. The clinical data cutoff date was 1 April 2019.
Results
Study population
Of the 29 enrolled PNH patients, 10 were complement treatment naive in part 2, and 19 were pretreated with eculizumab in part 3 (Table 1). Ten patients weighed >90 kg (3 and 7 patients in parts 2 and 3, respectively), a weight known to be associated with PD breakthrough with the SoC on label dose.6 In part 3, 4 patients (average weight, 110.7 kg) were on 1200 mg of eculizumab every 2 weeks because of a history of breakthrough on the label dose.
Demographic and baseline characteristics
| Characteristics | Eculizumab naive (part 2) IV loading doses on day 1 (375 mg), day 8 (500 mg), day 22 (1000 mg); 170 mg s.c. weekly starting on day 29 (N = 10) | Switched to crovalimab (part 3) | |||
|---|---|---|---|---|---|
| Group A: IV loading dose (1000 mg), followed by 680 mg s.c. every 4 wk* (n = 7) | Group B: IV loading dose (1000 mg), followed by 340 mg s.c. every 2 wk (n = 6) | Group C: IV loading dose (1000 mg), followed by 170 mg s.c. weekly (n = 6) | All (N = 19) | ||
| Age, mean ± SD, y | 53.9 ± 11.8 | 48.9 ± 4.8 | 53.2 ± 13.7 | 46.5 ± 13.7 | 49.5 ± 11.0 |
| Sex | |||||
| Female | 4 (40) | 4 (57.1) | 0 | 2 (33.3) | 6 (31.6) |
| Male | 6 (60) | 3 (42.9) | 6 (100) | 4 (66.7) | 13 (68.4) |
| Race | |||||
| Asian | 3 (30) | 2 (28.6) | 2 (33.3) | 3 (50.0) | 7 (36.8) |
| White | 7 (70) | 4 (57.1) | 3 (50.0) | 2 (33.3) | 9 (47.4) |
| Unknown | 0 | 1 (14.3) | 1 (16.7) | 1 (16.7) | 3 (15.8) |
| Weight, mean ± SD, kg | 75.35 ± 15.98 | 88.27 ± 28.36 | 85.67 ± 26.17 | 65.03 ± 19.65 | 80.11 ± 26.03 |
| C5 polymorphism | 1 (10) | 0 | 0 | 1 (17) | 1 (6) |
| Granulocyte clone size, mean ± SD | 80 ± 18 | 92 ± 8 | 84 ± 18 | 80 ± 13 | 86 ± 13 |
| Baseline LDH,† mean ± SD, U/L | 1159 ± 608 | 230 ± 46 | 415 ± 256 | 353 ± 323 | 327 ± 233 |
| Baseline hemoglobin, mean ± SD, g/L | 95.2 ± 14.7 | 106.4 ± 14.7 | 94.8 ± 8.3 | 112.5 ± 24.5 | 104.7 ± 17.7 |
| Eculizumab dose regimen before switch | |||||
| Labeled dose (900 mg every 2 wk) | NA | 5 (71) | 4 (67) | 6 (100) | 15 (79) |
| Higher dose or higher frequency (1200 mg every 2 wk) | NA | 2 (29) | 2 (33) | 0 (0) | 4 (21) |
| Patients with RBC transfusion in past 12 mo | 3 (30) | 3 (43) | 3 (50) | 2 (33) | 8 (42) |
| RBC units transfused in past 12 mo, n | 6 | 22 | 30 | 4 | 56 |
| Characteristics | Eculizumab naive (part 2) IV loading doses on day 1 (375 mg), day 8 (500 mg), day 22 (1000 mg); 170 mg s.c. weekly starting on day 29 (N = 10) | Switched to crovalimab (part 3) | |||
|---|---|---|---|---|---|
| Group A: IV loading dose (1000 mg), followed by 680 mg s.c. every 4 wk* (n = 7) | Group B: IV loading dose (1000 mg), followed by 340 mg s.c. every 2 wk (n = 6) | Group C: IV loading dose (1000 mg), followed by 170 mg s.c. weekly (n = 6) | All (N = 19) | ||
| Age, mean ± SD, y | 53.9 ± 11.8 | 48.9 ± 4.8 | 53.2 ± 13.7 | 46.5 ± 13.7 | 49.5 ± 11.0 |
| Sex | |||||
| Female | 4 (40) | 4 (57.1) | 0 | 2 (33.3) | 6 (31.6) |
| Male | 6 (60) | 3 (42.9) | 6 (100) | 4 (66.7) | 13 (68.4) |
| Race | |||||
| Asian | 3 (30) | 2 (28.6) | 2 (33.3) | 3 (50.0) | 7 (36.8) |
| White | 7 (70) | 4 (57.1) | 3 (50.0) | 2 (33.3) | 9 (47.4) |
| Unknown | 0 | 1 (14.3) | 1 (16.7) | 1 (16.7) | 3 (15.8) |
| Weight, mean ± SD, kg | 75.35 ± 15.98 | 88.27 ± 28.36 | 85.67 ± 26.17 | 65.03 ± 19.65 | 80.11 ± 26.03 |
| C5 polymorphism | 1 (10) | 0 | 0 | 1 (17) | 1 (6) |
| Granulocyte clone size, mean ± SD | 80 ± 18 | 92 ± 8 | 84 ± 18 | 80 ± 13 | 86 ± 13 |
| Baseline LDH,† mean ± SD, U/L | 1159 ± 608 | 230 ± 46 | 415 ± 256 | 353 ± 323 | 327 ± 233 |
| Baseline hemoglobin, mean ± SD, g/L | 95.2 ± 14.7 | 106.4 ± 14.7 | 94.8 ± 8.3 | 112.5 ± 24.5 | 104.7 ± 17.7 |
| Eculizumab dose regimen before switch | |||||
| Labeled dose (900 mg every 2 wk) | NA | 5 (71) | 4 (67) | 6 (100) | 15 (79) |
| Higher dose or higher frequency (1200 mg every 2 wk) | NA | 2 (29) | 2 (33) | 0 (0) | 4 (21) |
| Patients with RBC transfusion in past 12 mo | 3 (30) | 3 (43) | 3 (50) | 2 (33) | 8 (42) |
| RBC units transfused in past 12 mo, n | 6 | 22 | 30 | 4 | 56 |
Unless otherwise noted, all data are n (%).
NA, not applicable; s.c., subcutaneously; SD, standard deviation.
After the first 2 patients, the treatment regimen was modified to 170 mg weekly for the first 8 wk of subcutaneous dosing.
Upper limit of normal, 210 U/L.
One patient in each part was heterozygous for C5 c.2654G→A. All eligible patients rolled over into the OLE, with the exception of 1 patient in part 3 because of distance from the trial site and the inability to self-administer the drug. Two patients left the OLE at 35 and 44 weeks due to reasons not known to the sponsor, and 3 patients continued in part 3 at data cutoff.
Of the 25 patients in OLE, all patients in part 2 and 11 of 15 patients in part 3 were self-administering crovalimab outside of a clinical setting. See supplemental Figure 3 for patient disposition.
Safety
Overall, 145 AEs were observed in the 27 of 29 (93.1%) patients receiving crovalimab in parts 2 and 3 (Table 2). Nineteen of 145 AEs were considered related to treatment, and they were observed in 8 (27.6%) patients. All-causality AEs, occurring in ≥10% of patients were nasopharyngitis (31.0%), upper respiratory tract infection (24.1%), headache (17.2%), diarrhea (13.8%), arthralgia (10.3%), asthenia (10.3%), back pain (10.3%), pyrexia (10.3%) and urticaria (10.3%). Only 2 related AEs occurred in >1 patient: headache in 3 (10.3%) patients and urticaria in 2 (6.9%) patients. Six serious AEs (SAEs; atrial fibrillation, coronary artery stenosis, abdominal pain, bile duct stone, hemolysis, and nephrolithiasis) were reported in 5 (17.2%) patients. No treatment-related SAE or AE led to discontinuation of treatment. Two patients (6.9%) on 680 mg, every 4 weeks, in part 3 developed skin reactions that were considered related to treatment and possibly a manifestation of DTDCs. Both cases were mild or moderate in severity, transient, and resolved within ∼3 weeks while continuing crovalimab treatment. No injection site reaction, such as rash, induration, or injection pain, was reported. The duration of the injection is a few seconds.
Treatment-emergent AEs in patients with PNH
| Part 2: eculizumab naive (n = 10) | Part 3: switched to crovalimab (n = 19) | Part 2 + part 3 (N = 29) | ||||
|---|---|---|---|---|---|---|
| Duration of exposure period, median (range), wk | 83.5 (40-97) | 37.1 (13-69) | NA | |||
| Total patients with ≥1 AE, n (%) | 10 (100.0) | 17 (89.5) | 27 (93.1) | |||
| Total patients with ≥1 related AE, n (%) | 1 (10.0) | 7 (36.8) | 8 (27.6) | |||
| Total AEs, n | 40 | 105 | 145 | |||
| Total related AEs, n | 2 | 17 | 19 | |||
| Most common AEs,* no. of participants (%) | All causality | Related | All causality | Related | All causality | Related |
| Nasopharyngitis | 3 (30.0) | 0 | 6 (31.6) | 0 | 9 (31.0) | 0 |
| Upper respiratory tract infection | 4 (40.0) | 0 | 3 (15.8) | 0 | 7 (24.1) | 0 |
| Headache | 2 (20.0) | 1 (10.0) | 3 (15.8) | 2 (10.5) | 5 (17.2) | 3 (10.3) |
| Diarrhea | 0 | 0 | 4 (21.1) | 0 | 4 (13.8) | 0 |
| Arthralgia | 0 | 0 | 3 (15.8) | 1 (5.3) | 3 (10.3) | 1 (3.4) |
| Asthenia | 0 | 0 | 3 (15.8) | 1 (5.3) | 3 (10.3) | 1 (3.4) |
| Back pain | 2 (20.0) | 0 | 1 (5.3) | 0 | 3 (10.3) | 0 |
| Pyrexia | 1 (10.0) | 0 | 2 (10.5) | 0 | 3 (10.3) | 0 |
| Urticaria | 1 (10.0) | 1 (10.0) | 2 (10.5) | 1 (5.3) | 3 (10.3) | 2 (6.9) |
| Abdominal pain | 2 (20.0) | 0 | 0 | 0 | 2 (6.9) | 0 |
| Bronchitis | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Dizziness | 0 | 0 | 2 (10.5) | 1 (5.3) | 2 (6.9) | 1 (3.4) |
| Exertional dyspnea | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Dysuria | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Hemolysis | 0 | 0 | 2 (10.5) | 1 (5.3) | 2 (6.9) | 1 (3.4) |
| Influenza | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Vertigo | 2 (20.0) | 0 | 0 | 0 | 2 (6.9) | 0 |
| Patients with ≥1 SAE, n (%) | 3 (30.0) | 0 | 2 (10.5) | 0 | 5 (17.2) | 0 |
| AE leading to discontinuation of treatment, n | 0 | 0 | 0 | 0 | 0 | 0 |
| Part 2: eculizumab naive (n = 10) | Part 3: switched to crovalimab (n = 19) | Part 2 + part 3 (N = 29) | ||||
|---|---|---|---|---|---|---|
| Duration of exposure period, median (range), wk | 83.5 (40-97) | 37.1 (13-69) | NA | |||
| Total patients with ≥1 AE, n (%) | 10 (100.0) | 17 (89.5) | 27 (93.1) | |||
| Total patients with ≥1 related AE, n (%) | 1 (10.0) | 7 (36.8) | 8 (27.6) | |||
| Total AEs, n | 40 | 105 | 145 | |||
| Total related AEs, n | 2 | 17 | 19 | |||
| Most common AEs,* no. of participants (%) | All causality | Related | All causality | Related | All causality | Related |
| Nasopharyngitis | 3 (30.0) | 0 | 6 (31.6) | 0 | 9 (31.0) | 0 |
| Upper respiratory tract infection | 4 (40.0) | 0 | 3 (15.8) | 0 | 7 (24.1) | 0 |
| Headache | 2 (20.0) | 1 (10.0) | 3 (15.8) | 2 (10.5) | 5 (17.2) | 3 (10.3) |
| Diarrhea | 0 | 0 | 4 (21.1) | 0 | 4 (13.8) | 0 |
| Arthralgia | 0 | 0 | 3 (15.8) | 1 (5.3) | 3 (10.3) | 1 (3.4) |
| Asthenia | 0 | 0 | 3 (15.8) | 1 (5.3) | 3 (10.3) | 1 (3.4) |
| Back pain | 2 (20.0) | 0 | 1 (5.3) | 0 | 3 (10.3) | 0 |
| Pyrexia | 1 (10.0) | 0 | 2 (10.5) | 0 | 3 (10.3) | 0 |
| Urticaria | 1 (10.0) | 1 (10.0) | 2 (10.5) | 1 (5.3) | 3 (10.3) | 2 (6.9) |
| Abdominal pain | 2 (20.0) | 0 | 0 | 0 | 2 (6.9) | 0 |
| Bronchitis | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Dizziness | 0 | 0 | 2 (10.5) | 1 (5.3) | 2 (6.9) | 1 (3.4) |
| Exertional dyspnea | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Dysuria | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Hemolysis | 0 | 0 | 2 (10.5) | 1 (5.3) | 2 (6.9) | 1 (3.4) |
| Influenza | 0 | 0 | 2 (10.5) | 0 | 2 (6.9) | 0 |
| Vertigo | 2 (20.0) | 0 | 0 | 0 | 2 (6.9) | 0 |
| Patients with ≥1 SAE, n (%) | 3 (30.0) | 0 | 2 (10.5) | 0 | 5 (17.2) | 0 |
| AE leading to discontinuation of treatment, n | 0 | 0 | 0 | 0 | 0 | 0 |
AEs are presented as Medical Dictionary for Regulatory Activities (v21.1) Preferred Terms. AE counts are per individual, and multiple occurrences of the same event in an individual are counted only once.
Most common AEs include AEs with a frequency ≥2 in part 2 or 3.
Pharmacokinetics
Bioavailability was estimated at 100%, exposure was dose proportional within the dose range investigated, and terminal half-life was estimated at ∼30 days for a 70-kg patient. Elimination of crovalimab was faster in patients who switched to crovalimab than in treatment-naive patients and HVs. This effect was transient and decreased over time. Steady-state concentration levels were reached at around days 200 and 300 in parts 2 and 3, respectively (Figure 1).
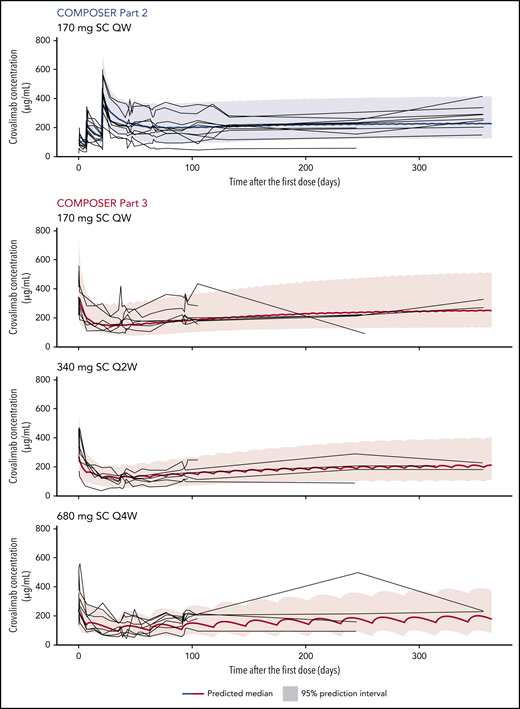
Individual (gray lines) PK data and modeled mean (bold blue and red lines) and 95% confidence interval for crovalimab concentration over time in PNH patients.
Individual (gray lines) PK data and modeled mean (bold blue and red lines) and 95% confidence interval for crovalimab concentration over time in PNH patients.
As predicted, DTDCs of crovalimab, C5, and eculizumab of different sizes were detected in all switch patients but not in any of the treatment-naive patients (negative control) using size-exclusion chromatography with a crovalimab-specific ELISA. The complexed crovalimab showed an initial peak, with a predominance of mid-to-high molecular weight DTDCs on day 15, followed by a steady decrease; complexes were undetectable from day 78 onward.
Pharmacodynamics
Crovalimab completely blocked the terminal complement pathway activity (<10 U/mL) in all treatment-naive patients and patients who switched to crovalimab (Figure 2A). In part 2, the terminal complement pathway was already fully inhibited on day 9, after the second uptitration dose of 500 mg of crovalimab. In part 3, 7 of the 19 included patients had LIA values >10 U/mL on day −14 and/or day −1 precrovalimab, showing incomplete inhibition of the terminal complement pathway while on eculizumab. One of these 7 patients had the C5 mutation; the other 6 patients exhibited LIA values of 11 to 59 U/mL and had a mean weight of 102.2 kg (range, 89-123).
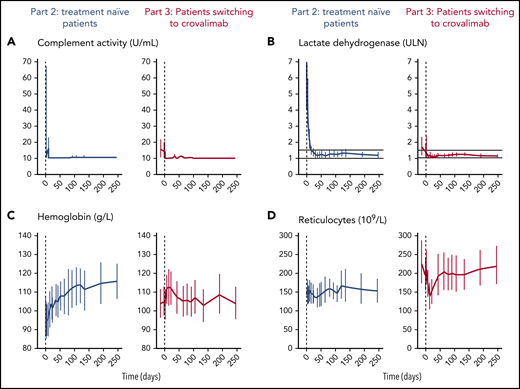
PD markers, hemoglobin, and reticulocyte count over time in PNH patients. Mean and 95% CI for complement activity by LIA (A), LDH (normalized) (B), hemoglobin (C), and reticulocytes (D).
PD markers, hemoglobin, and reticulocyte count over time in PNH patients. Mean and 95% CI for complement activity by LIA (A), LDH (normalized) (B), hemoglobin (C), and reticulocytes (D).
tC5 serum concentration
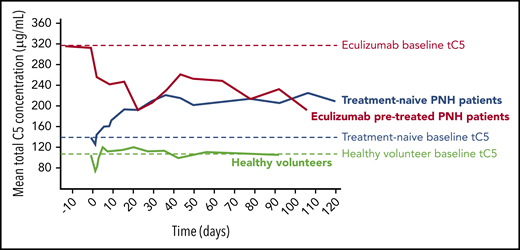
In the HVs enrolled in part 1 of this study, mean tC5 serum concentration was 103.14 µg/mL (standard deviation 19.43 µg/mL) before crovalimab administration (Figure 3). Treatment-naive patients in part 2 had a slightly higher mean baseline tC5 concentration compared with HVs (133.69 ± 38.41 µg/mL). tC5 increased after administration of crovalimab and plateaued at 217.60 ± 42.87 µg/mL at week 12. Mean baseline tC5 was markedly higher in patients pretreated with eculizumab (316.76 ± 75.60 µg/mL) compared with HVs and treatment-naive patients. Once treated with crovalimab, mean tC5 decreased considerably over the following weeks to reach a lower plateau level that was indistinguishable from that in treatment-naive patients after 12 weeks (203.74 ± 65.26 µg/mL) (Figure 3).
fC5
In treatment-naive patients, median fC5 decreased to 14.60 ng/mL (range, 8.9-34.2) after the first IV dose of 375 mg and, after a small increase, remained very low thereafter (supplemental Figure 7; 10.25 ng/mL; range, 6.3-16.0 at week 12). In patients switching to crovalimab, median fC5 was 12.85 ng/mL (range, 7.83-110.3) at day 2 and 12.61 ng/mL (range, 5.2-23.1) at week 12.
Immunogenicity
ADAs were detected in 5 of 10 (50%) treatment-naive patients and in 3 of 19 (17%) patients switching to crovalimab. One treatment-naive patient with ADAs experienced moderate PK reduction (∼35%) and a slight increase in LDH (1.37× upper limit of normal [ULN]) at week 14. LDH returned to lower values at week 36 (1.13× ULN), and the patient remained clinically stable with no safety event. No impact of ADAs was seen in PK/PD and safety profiles in the remaining ADA-positive patients.
Efficacy
LDH
Treatment-naive patients in part 2 showed immediate sustained LDH reduction responses, with 9 of 10 patients achieving LDH <1.5× ULN after 16 weeks (Figure 2B; supplemental Figure 4).
In part 3, 6 of 19 patients (32%) had LDH values > 1.5× ULN on day −14 and/or day −1 precrovalimab while still on eculizumab, with a mean weight of 80.2 kg (range, 51-102). LDH levels in these 6 patients decreased to <1.5× ULN following crovalimab treatment. The remaining patients had stable or reduced LDH (Figure 2C; supplemental Figure 5). The 2 Japanese patients with the C5 mutation had an LDH response like other patients without mutations, indicating full suppression of terminal complement pathway activity (supplemental Figure 6).
Hemoglobin, reticulocyte count, and transfusion need
Mean hemoglobin values for treatment-naive patients increased from 95.2 g/L (standard deviation, 14.7) to 116.0 g/L (standard deviation, 16.8) at week 16 (Figure 2C, left panel). In pretreated patients, hemoglobin was stable over the observation period (Figure 2C, right panel). No major change in reticulocyte count was observed in either group (Figure 2D).
In part 2, 3 of 10 patients had received blood transfusions during the 12 months preceding the study; 2 of them became transfusion-free while on crovalimab, whereas the third patient remained on transfusions (supplemental Table 1). One patient who did not have transfusions previously required 2 transfusions of 2 units each while on crovalimab. Hemoglobin concentration in the 8 patients not requiring transfusions while on crovalimab increased by 16 g/L (range, 4-37) from baseline to week 16 (supplemental Figure 4).
Of the 19 patients in part 3, 8 patients received ≥1 RBC transfusion in the 12 months prior to crovalimab, and 5 patients received ≥1 unit while on crovalimab (supplemental Table 1). Overall transfusion requirements seemed to be slightly reduced with crovalimab. Beyond the first year of follow-up, 1 previously transfusion-free patient required 1 transfusion (2 units) during an infection on study day 400.
Overall, 2 patients received additional IV doses of crovalimab; 1 because of BTH, as judged by the investigator, and 1 for fatigue (the latter in the absence of formal BTH, according to our definition). Both patients improved and continued to receive crovalimab according to their treatment schedule.
Mean hemoglobin and mean reticulocyte count, with 95% confidence intervals (CIs), are shown in Figure 2C and D, respectively. Individual hemoglobin and transfusion need are shown in supplemental Figures 4 and 5.
BTH
No patient in part 2 met the BTH criteria or had BTH reported as an AE. In part 3, 4 of 19 patients met the criteria for BTH; 1 was reported as an SAE with concomitant viral infection. The other 3 events were not reported as AE “breakthrough,” and concomitant otitis media or nasopharyngitis was reported in 2 cases. One patient had elevated LDH with anemia and no AE reported. With a cumulative observation period of 29.9 years, this corresponds to 0.13 BTH per year of treatment (95% CI, 0.04-0.34).
One AE was reported as “BTH” by the investigator, but it did not meet the post hoc criteria for BTH, because LDH was <2× ULN.
HRQoL assessments
Patients in part 2 experienced a mean improvement of 8.8 points on the FACIT-Fatigue questionnaire from baseline to week 10; EORTC QLQ-C30 scores also improved by an average of 21.1 points. Mean improvements on both scales exceeded the cutoff for clinically meaningful improvement of ≥3 points14,15 and ≥10 points,16 respectively. Additional mean improvements of 17.3 and 19.2 points were observed on physical functioning and global-health-status/QoL-scales, respectively. The scales demonstrated a consistent pattern of change across individuals.
Patients in part 3 maintained baseline fatigue levels on the FACIT-Fatigue and EORTC QLQ-C30 scales up to week 12. Neither the physical functioning nor the global health status/QoL scales demonstrated a clinically meaningful change.
Discussion
Crovalimab, administered subcutaneously in small volumes (1-4 mL) at intervals of up to 4 weeks, showed excellent tolerability and acceptable safety while delivering sustained complete terminal complement pathway inhibition.
Crovalimab targets a different epitope on C5 than the approved compounds; therefore, it may serve a more complete patient population with greater efficacy. The broad inclusion criteria of part 3, allowing patients with recent transfusion, elevated LDH, recent history of BTH, or higher-than-label dose eculizumab, resulted in an enrichment of patients in the study population who had a relatively high body weight, needed more transfusions, and, in some cases, were on escalated doses of eculizumab. This population is likely to better reflect the general PNH population on C5 inhibitor treatment17,18 and contrasts with the population of registration trials for ravulizumab that focused on well-controlled patients with relatively low average body weight.2 Specifically, 11 of 19 patients in part 3 were on higher-than-label eculizumab dose, had LDH >1.5× ULN on eculizumab, and/or had residual clinically relevant complement activation at the trough of eculizumab, as indicated by LIA >10 U/mL.2,5
The 4 patients in part 3 who were treated with 1200 mg of eculizumab (2 of 4 requiring occasional transfusion) before switching to crovalimab were stably maintained at any treatment interval on crovalimab. One of the 2 transfusion-dependent patients on 1200 mg of eculizumab became transfusion independent on an every-4-week schedule of crovalimab. Taken together, these data indicate that 680 mg of crovalimab, subcutaneously every 4 weeks, is likely to be an effective and safe dose regimen with relatively low treatment burden, even for patients on escalated doses of eculizumab that are not well controlled.
Moreover, crovalimab treatment resulted in clinically meaningful improvements in fatigue, physical functioning, and global health status/QoL, on average, and reflected the sustained and effective suppression of complement activation, comparing favorably with results reported for other C5 inhibitors in treatment-naive patients.1,3,13
An excess of C5 binding sites has been reported to correlate with low incidence of BTH.19 Using SMART, the accumulation of tC5 was limited, and eculizumab-pretreated patients experienced a major drop in tC5 (Figure 3). This is indicative of the C5-disposing properties of crovalimab, resulting in crovalimab/C5 ratios >1:2. This can be achieved and maintained throughout the dosing interval with small amounts of antibody in easily injectable volumes (1-2 × 2 mL) delivered subcutaneously within seconds.
The annualized breakthrough event rate across COMPOSER parts 2 and 3 is 0.13 (95% CI, 0.04-0.34; supplemental Table 2). This compares to BTH event rates of 0 to 0.14 (ravulizumab) and 0.06 to 0.46 (eculizumab), as reported in recent phase 3 studies.2,3 Given that these were conducted in well-controlled, label-dose, and moderate weight patient populations, crovalimab seems to have a favorable activity with regard to BTH event rate. It must be noted that BTH analysis was introduced post hoc, possibly limiting the comparability with pivotal trials of ravulizumab.2,3
A limitation of the study is the lack of a randomized design and a formal comparison arm. Our study uses a design in part 3 to allow for a switchover comparison on an individual patient level. Crovalimab was specifically engineered to provide a self-administered treatment option for PNH patients based on the well-established C5 target. The targeted steps to improve the biology of the antibody resulted in an unparalleled low amount of drug needed. Crovalimab, in comparison with the currently approved C5 antibodies, can be used in lower doses (eg, 1360 mg of crovalimab per 8 weeks vs 3600-4800 mg of eculizumab or 3000-3600 mg of ravulizumab), depending on the patient’s weight. Combined with the C5-disposing properties, crovalimab is the only C5-targeting drug that can stably control an “all-comer” population of PNH patients, including the C5 SNP.
The COMPOSER study showed that crovalimab was well tolerated without clinically relevant injection site reactions or immunogenicity, leading to complete terminal complement pathway inhibition, in all patients. Transient DTDC formation in part 3 patients, resulting from the concurrent systemic presence of eculizumab and crovalimab, did not result in undue safety events or clinically relevant loss of efficacy. SMART-engineered crovalimab allows infrequent low-volume fast subcutaneous self-administration, offering PNH patients treatment that is independent of clinical settings. These promising results support the continued development of crovalimab in PNH.
Data sharing requests should be sent to Alexander Röth (alexander.roeth@uk-essen.de).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sasha Srekovich, Ido Paz-Priel, Anna Kiialainen, and Charlotte Vignal for helpful discussions, Jules Hernandez-Sanchez for statistical support, Niels Janssen, Erica Winter, Katrijn Bogman, and Joy Hsu for valuable contributions during start up and conduct of the trial, Elena Fernandez for continuous support, Margarida Duarte Goncalves for never-ending energy and leadership, and all patients for compliance with a very demanding schedule of assessment that enabled this trial.
This work was supported by F. Hoffmann-La Roche Ltd and Chugai Pharmaceutical.
Authorship
Contribution: B.K. and C.B. developed the protocol, analyzed and interpreted the data, and wrote the manuscript; A.J. and K.S. developed the protocol, analyzed and interpreted the data, and contributed to the manuscript; A.R. contributed to the development of the protocol, recruited patients, collected data, analyzed and interpreted the data, and contributed to the manuscript; J.-i.N. and R.P.d.L. recruited patients, collected, analyzed, and interpreted the data, and contributed to the manuscript; A.S., A.D., B.G., G.J., J.H., and J.A.-C. analyzed and interpreted the data, and contributed to the manuscript; M.B.-S. interpreted the data and contributed to the manuscript; F.S.d.F., H.S., J.G.-W., J.S., K.U., J.S.K., M.E., H.N., J.P., S.I., S.S., S.-S.Y., Y.I., and Z.N. recruited patients and contributed to the manuscript; and all authors approved the final version.
Conflict-of-interest disclosure: A.S., A.D., J.G.-W., J.A.-C., and M.B.-S. are employed by Roche. B.K., and C.B. are employed by and own shares of Roche. J.H. is a former employee of Roche. H.S. has received research support, acted as a consultant for, and received honoraria from Alexion Pharmaceuticals and Novartis and has acted as a consultant for Roche (all to University Hospital Ulm). B.G. is employed by Genentech. A.J. is a former employee of, and owns stock in, Genentech. K.S. is employed by Chugai Pharmaceuticals. A.R. has received honoraria, consulting fees, and research support from Alexion Pharmaceuticals, Novartis, and Roche. F.S.d.F. has received research funding and honoraria and has acted as a consultant for Alexion Pharmaceuticals and Novartis. J.N. is a member of the advisory board for Chugai Pharmaceuticals and Alexion Pharmaceuticals and has received research funding and honorarium from Alexion Pharmaceuticals. K.U. has received research funding from Alexion Pharmaceuticals and Chugai Pharmaceuticals and is a member of the speakers bureau for Chugai Pharmaceuticals. J.P. has acted as a consultant for and received honoraria from Novartis, Alexion Pharmaceuticals, Amgen, Roche, Pfizer Inc., and Boehringer Ingelheim. R.P.d.L. has received research funding from Alexion Pharmaceuticals, Pfizer, Novartis, and Amgen, has received honoraria from Alexion Pharmaceuticals, Pfizer, and Novartis, and has acted as a consulted for Alexion Pharmaceuticals, Pfizer, Novartis, and Roche. The remaining authors declare no competing financial interests.
The current affiliation for J.H. is Genmab A/S, Copenhagen, Denmark.
The current affiliation for A.J. is Gilead Sciences, Foster City, CA.
Correspondence: Alexander Röth, University Hospital Essen, West German Cancer Center, Department of Hematology, University Duisburg-Essen, Hufelandstrasse 55, 45147 Essen, Germany; e-mail: alexander.roeth@uk-essen.de.

Comments
Hemoglobinuria versus hematuria