


Abstract
Integrins are a large family of heterodimeric cell surface receptors that bind prototypic ligands on neighboring cells or in the extracellular matrix. Numerous studies have revealed key roles for platelet and leukocyte integrins in adhesion and migration and, thereby, their significance for hemostasis and immunity. The clinical importance of these integrins has also become clear, because aberrant integrin expression and/or behavior are associated with bleeding disorders, immunodeficiency, or autoimmune diseases. Importantly, overwhelming evidence gathered over recent years shows that regulation of integrin function is far more complex than previously assumed; a picture has emerged of multiple cytoplasmic, cell surface, and extracellular regulators working together to ensure cell type–specific and integrin-specific control of integrin functions. Here, we discuss recent insights into the dynamic activation and suppression of hematopoietic integrins, as well as their implications for platelet and leukocyte function in health and disease.
Integrins and their functions on hematopoietic cells
The integrins are a family of 24 transmembrane αβ heterodimeric glycoproteins that are assembled from 18 α-subunits and 8 β-subunits. Integrins bind to proteins in the extracellular matrix (ECM) or on the surface of other cells in a metal ion–dependent manner, while intracellularly connecting to contractile actin filaments and multiple signaling pathways. Thus, integrins stimulate dynamic adhesive cell–cell or cell-ECM interactions, as well as cell migration, proliferation, and survival.1 On hematopoietic cells, integrins regulate leukocyte activation and transmigration, immunological synapse formation, platelet aggregation, and phagocytosis (Figure 1A-B).1-4

Integrins and their functions on hematopoietic cells. (A) The human integrin family consists of 24 heterodimers, composed of 18 α-subunits and 8 β-subunits; underscored are those subunits of which the heterodimer has not been detected on hematopoietic cells (α6β4, αvβ6, α7-11β1). The matching colors represent exemplary heterodimers together with a prototypic ligand and the biological process in which this receptor/ligand pair is involved (eg, the prototypic ligand of α4β1 is VCAM-1, which is involved in leukocyte extravasation). Integrins support diverse events in the hematopoietic life cycle, including maintenance of progenitors in the bone marrow niche, B- and T-cell proliferation, maturation and differentiation in the thymus and spleen, and homing/retention of tissue-resident leukocytes in the epithelia of the gut and skin. In addition, integrins are required for hemostasis, transforming growth factor-β activation, migration in and out of blood vessels or secondary lymphoid tissues, and migration through environments rich in collagens, laminins, and fibronectin. (B) Integrins also promote leukocyte effector functions by potentiating the formation of immunological synapses and cytokine production, as well as phagocytosis of apoptotic cells and (complement-opsonized) pathogens. (C) Integrins adopt 3 main conformations on the cell surface, with different affinities (low, intermediate, or high) for ligand. MadCAM, mucosal addressin cell adhesion molecule; MHC, major histocompatibility complex; SMAC, supramolecular activation cluster; TGF, transforming growth factor; VCAM, vascular cell adhesion molecule.
Integrins and their functions on hematopoietic cells. (A) The human integrin family consists of 24 heterodimers, composed of 18 α-subunits and 8 β-subunits; underscored are those subunits of which the heterodimer has not been detected on hematopoietic cells (α6β4, αvβ6, α7-11β1). The matching colors represent exemplary heterodimers together with a prototypic ligand and the biological process in which this receptor/ligand pair is involved (eg, the prototypic ligand of α4β1 is VCAM-1, which is involved in leukocyte extravasation). Integrins support diverse events in the hematopoietic life cycle, including maintenance of progenitors in the bone marrow niche, B- and T-cell proliferation, maturation and differentiation in the thymus and spleen, and homing/retention of tissue-resident leukocytes in the epithelia of the gut and skin. In addition, integrins are required for hemostasis, transforming growth factor-β activation, migration in and out of blood vessels or secondary lymphoid tissues, and migration through environments rich in collagens, laminins, and fibronectin. (B) Integrins also promote leukocyte effector functions by potentiating the formation of immunological synapses and cytokine production, as well as phagocytosis of apoptotic cells and (complement-opsonized) pathogens. (C) Integrins adopt 3 main conformations on the cell surface, with different affinities (low, intermediate, or high) for ligand. MadCAM, mucosal addressin cell adhesion molecule; MHC, major histocompatibility complex; SMAC, supramolecular activation cluster; TGF, transforming growth factor; VCAM, vascular cell adhesion molecule.
Integrins can adopt different conformational states regulating their affinity for ligands. At least 3 primary conformations are distinguished, including the low-affinity “bent” conformation, the intermediate-affinity “extended conformation with closed headpiece,” and the high-affinity “extended conformation with open headpiece” (Figure 1C).4 Most platelet and leukocyte integrins assume the bent conformation by default, to prevent interactions with ligands and, thus, cell adhesion.4 The bent conformation is energetically favorable and is maintained by a “clasp” formed by the membrane-proximal GFFKR region in the α-cytoplasmic tail and the HDRxE region in the β-tail (Figure 2). The clasp is stabilized by a salt bridge in certain integrins, such as αIIbβ3, the main integrin on platelets.4 Upon extracellular (outside-in) or intracellular (inside-out) signals, integrins rapidly undergo a conformational change toward the extended high-affinity conformation, a process that is called “integrin activation” and promotes ligand binding.
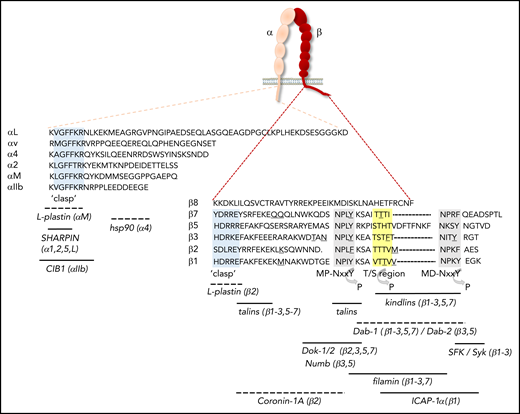
Integrin cytoplasmic tails and binding sites for proteins that regulate activation. The amino acid sequences of the cytoplasmic tails of human αL, α2, α4, αIIb, αv, and αM, as well as of β1-3, β5, β7, and β8, are shown. With the exception of the “clasp” region, the cytoplasmic tails of α-subunits differ considerably in sequence and length, whereas a number of conserved motifs in β-cytoplasmic tails are identified. Gray shading indicates the membrane-proximal (MP) and membrane-distal (MD) NxxY motifs, yellow shading denotes the T/S region, and light blue shading demarcates the α-tail GFFKR and β-tail HDRxE sequences that form the “clasp” to stabilize the bent conformation. Underscored residues indicate sites of calpain cleavage, whereas regions that can be phosphorylated are indicated by an arrow. Binding sites for regulators of integrin activation are indicated, with the integrin subunits in parentheses indicating those to which binding has been shown. Dashed lines indicate that the exact binding site has not been mapped. CIB1, calcium- and integrin-binding protein 1; hsp, heat shock protein; SFK, Src-family kinases.
Integrin cytoplasmic tails and binding sites for proteins that regulate activation. The amino acid sequences of the cytoplasmic tails of human αL, α2, α4, αIIb, αv, and αM, as well as of β1-3, β5, β7, and β8, are shown. With the exception of the “clasp” region, the cytoplasmic tails of α-subunits differ considerably in sequence and length, whereas a number of conserved motifs in β-cytoplasmic tails are identified. Gray shading indicates the membrane-proximal (MP) and membrane-distal (MD) NxxY motifs, yellow shading denotes the T/S region, and light blue shading demarcates the α-tail GFFKR and β-tail HDRxE sequences that form the “clasp” to stabilize the bent conformation. Underscored residues indicate sites of calpain cleavage, whereas regions that can be phosphorylated are indicated by an arrow. Binding sites for regulators of integrin activation are indicated, with the integrin subunits in parentheses indicating those to which binding has been shown. Dashed lines indicate that the exact binding site has not been mapped. CIB1, calcium- and integrin-binding protein 1; hsp, heat shock protein; SFK, Src-family kinases.
Here, we review recent insights into how hematopoietic integrins are dynamically activated and importantly, inactivated, by cytoplasmic proteins or by other transmembrane receptors and extracellular cues.
Regulation of integrin activation
Integrin conformations and the canonical pathway to inside-out activation
Integrin inside-out activation is tightly regulated by an intracellular sequence of events initiated by signals from several receptors. In leukocytes, these include G protein–coupled receptors activated by chemokines or inflammatory cytokines (Figure 3A), as well as the B-cell receptor or T-cell receptor (TCR) activated by antigens.1-4 In platelets, inside-out activation is triggered by a variety of receptors recognizing soluble agonists or immobilized ligands in the subendothelial matrix that are exposed upon vascular injury (Figure 3B).4 From a large body of literature, a “canonical” pathway to integrin activation has emerged, primarily involving the generation of the second messengers Ca2+ and diacylglycerol, together activating protein kinase C and the GTPase Rap1 (Figure 3). Rap1 subsequently binds the effector protein Rap1-guanosine triphosphate (GTP)–interacting adaptor molecule (RIAM), which recruits the cytoplasmic protein talin-1 to the plasma membrane (Figure 3A).4,5 RIAM disrupts an autoinhibitory conformation of talin-1, allowing the FERM domain in the N-terminal talin head to interact with membrane phospholipids and 2 regions in the integrin β-cytoplasmic tail (Figure 3A).4 In turn, rod domains in the talin tail interact with actin filaments and increase integrin avidity by facilitating integrin clustering through the formation of antiparallel talin homodimers (Figure 3A).5 Intriguingly, talin can switch between a compacted conformation that simultaneously prevents talin binding to phospholipids, integrin tails, and actin, and an unfolded string-like conformation that allows these interactions.6 In addition to talin, a FERM domain–containing protein of the kindlin family is recruited to bind an additional site in the β-subunit; the coordinated talin/kindlin binding results in unclasping of the α- and β-tails, causing a conformational change that is propagated across the membrane toward the high-affinity state (Figure 3A).5 Although humans express 3 kindlin family members, kindlin-3 is restricted to hematopoietic cells; mutations in the gene encoding kindlin-3 cause leukocyte adhesion deficiency III, a combined disorder characterized by immunodeficiency and bleeding as a result of impaired β1, β2, and β3 integrin activation on leukocytes and platelets.7-11 The recruitment of components of the activation pathway, including talin-1, kindlin-3, and the Rap1/RIAM complex in T cells and neutrophils, is also dependent on Src kinase–associated phosphoprotein and adhesion and degranulation-promoting adaptor protein, at least in response to some stimuli.12,13 More recently, the actin-binding protein coronin-1A, which is required for innate and adaptive immunity, was also shown to increase activation of αLβ2 integrins, which involves direct interactions with the β2-cytoplasmic tail (Figure 2).14 Integrin β-subunits contain a number of conserved sequences, commonly referred to as the membrane-proximal and membrane-distal NxxY motifs and the intervening threonine/serine (T/S) region, which bind a variety of integrin-associated proteins, including talins and kindlins (Figure 2). The cytoplasmic tails of α-subunits are generally much more diverse (Figure 2), allowing for α-subunit–specific interactions. For example, calcium- and integrin-binding protein-1 interacts with the αIIb tail and is important for platelet activation, whereas heat shock protein 90 (hsp90) binds to the α4-tail (Figure 2).15,16 hsp90 enhances talin-1/kindlin-3 interactions with β1/β7 tails, thus promoting inside-out activation of α4β1 and α4β7 integrins, and disruption of hsp90 binding prevents fever-induced T-cell transmigration in mice.16 Because hsp90 expression is strongly augmented by high fever, these data uncover a direct link among fever, integrin activation, and T-cell recruitment.
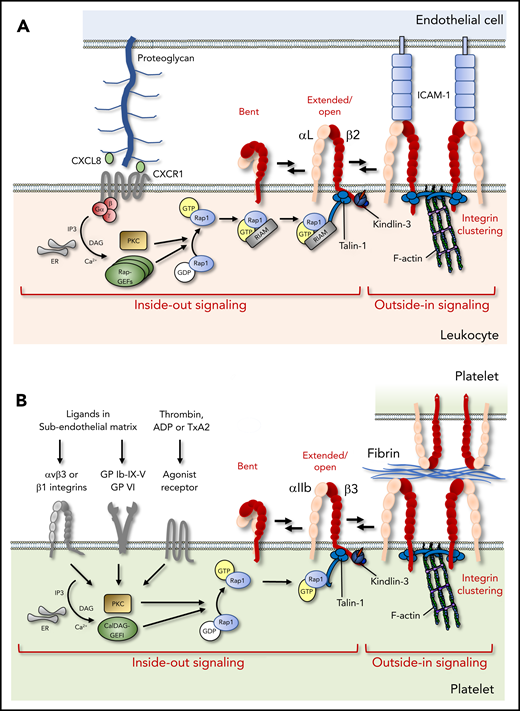
Integrin activation in leukocytes and platelets. (A) Chemokines presented on the endothelial surface activate leukocyte integrins via G protein–coupled receptors, which trigger Rap1 activation, RIAM-dependent talin-1 mobilization, and kindlin-3 recruitment to the integrin β-cytoplasmic tail, leading to the allosteric change toward the high-affinity conformation (inside-out signaling). Subsequent ligand binding, integrin clustering, and connection to the cytoskeleton ensure cell adhesion and amplify downstream signaling (outside-in signaling). (B) In platelets, soluble agonist or matrix receptors can trigger inside-out activation, which depends critically on CalDAG-GEFI. In contrast, RIAM is not required, and Rap1 can bind directly to talin-1. ADP, adenosine diphosphate; ER, endoplasmic reticulum; GDP, guanosine diphosphate; GP, glycoprotein, IP3, inositol trisphosphate; PKC, protein kinase C; TxA2, thromboxane A2.
Integrin activation in leukocytes and platelets. (A) Chemokines presented on the endothelial surface activate leukocyte integrins via G protein–coupled receptors, which trigger Rap1 activation, RIAM-dependent talin-1 mobilization, and kindlin-3 recruitment to the integrin β-cytoplasmic tail, leading to the allosteric change toward the high-affinity conformation (inside-out signaling). Subsequent ligand binding, integrin clustering, and connection to the cytoskeleton ensure cell adhesion and amplify downstream signaling (outside-in signaling). (B) In platelets, soluble agonist or matrix receptors can trigger inside-out activation, which depends critically on CalDAG-GEFI. In contrast, RIAM is not required, and Rap1 can bind directly to talin-1. ADP, adenosine diphosphate; ER, endoplasmic reticulum; GDP, guanosine diphosphate; GP, glycoprotein, IP3, inositol trisphosphate; PKC, protein kinase C; TxA2, thromboxane A2.
Variations to conformational regulation and inside-out activation mechanisms
In recent years, our knowledge of integrin regulation has greatly expanded, and a complex picture has emerged in which distinct integrins can behave very differently. Interestingly, the salt bridge between the α- and the β-tails seems dispensable for the regulation of most β1 integrins in vivo.17 The notable exception is α4β1, in which disruption of the salt bridge leads to hyperadhesion of leukocytes to endothelium, thereby preventing transmigration.17,18 Because β1 integrins on leukocytes are generally more important for the binding of ECM proteins in tissues/organs or during the later stages of transmigration, they probably require less robust activation than β2 and β3 integrins, which mediate firm cell adhesion under flow. Indeed, a variety of studies using soluble integrin ectodomains or intact integrins embedded in phospholiposomes clearly demonstrate that αXβ2 and αvβ3 assume strongly bent conformations in the absence of activating stimuli, whereas β1 integrins naturally adopt a variety of intermediate conformations, including half-bent, as well as extended/closed and extended/open, under these conditions.19-21 Thus, it is likely that affinity regulation in β1 integrins happens through more subtle allosteric rearrangements, involving the opening and closing of the headpiece domain, rather than by large-scale global changes. Most intriguingly, the cytoplasmic domain of integrin β8 is completely different from that of all other β-subunits; it seems unstructured and lacks conserved motifs (Figure 2).22 Therefore, this integrin cannot interact with talins or kindlins, and it was recently reported that αvβ8 is constitutively in an extended active conformation.23 In addition, its affinity for ligand is not strongly increased by Mn2+, unlike that of most other integrins.23,24 Thus, it is now clear that individual integrin heterodimers differentially tune their affinity for ligands.
In addition, it appears that distinct integrin-dependent processes also differ in their requirements for integrin-interacting proteins. For example, although talin-1 and kindlin-3 are necessary for efficient leukocyte adhesion to endothelium under flow, including T-cell homing to lymph nodes, kindlin-3 is dispensable for T-cell diapedesis, T-cell activation in the spleen, intrathymic T-cell migration and maturation, and T-cell release into the circulation.25-28 Moreover, T-cell extravasation from the inflamed microvasculature in the brain, as well as the onset of experimental autoimmune encephalitis (a murine model for multiple sclerosis), can occur in the absence of kindlin-3, whereas recruitment of autoreactive T cells to the central nervous system in normal conditions cannot.25 Finally, T-cell progenitor homing to the thymus requires kindlin-3 in postnatal or adult animals, but not during development when the thymus is still avascular.29 The model emerging from these data is that kindlin-3 is dispensable under low shear stress or when excess ligand is available, whereas talin-1 and kindlin-3 are both required under high shear stress or ligand-limiting conditions. A possible explanation for these results is provided by observations that talin-1 alone can induce the bent-to-extended change in integrin conformation, which leads to the largest increase in affinity, whereas kindlin-3 is only required for the headpiece opening and, therefore, only modestly enhances the affinity.30 In addition, it was recently found that kindlins can also form dimers and that they stimulate integrin clustering, thus further promoting integrin avidity and firm adhesion under flow.31-33
It has also become clear in recent years that there are several variations to the canonical pathway to integrin activation, which may depend on the cell type, integrin heterodimer, or activating stimulus. For instance, activation of Rap1 can occur by several guanine nucleotide exchange factors (GEFs), such as Ca2+- and DAG-regulated GEF1 (CalDAG-GEF1) (Figure 3B). Disruptive mutations in CalDAG-GEF1 in humans cause integrin-activation defects in platelets but not in leukocytes; therefore, these patients develop bleeding but not immunodeficiency.34,35 Possibly, residual expression of CalDAG-GEF1 precludes leukocyte defects, or other GEFs can compensate for the loss of function of CalDAG-GEF1. Indeed, other GEFs can also activate Rap1 in leukocytes and promote integrin activation, including RapGEF1, RapGEF3, and RapGEF6 (Figure 3A).2,4 Moreover, although abundant evidence clearly shows that talin-1 and Rap1 are absolutely required for correct function of all hematopoietic integrins, RIAM only seems to be required for talin recruitment to β2 integrins (Figure 3). Indeed, the loss of RIAM in mice completely prevented β2 integrin activation, leading to leukocyte defects, whereas α4β1 activation was only partially inhibited, and αIIbβ3 was not affected at all.36-38 RIAM is expressed at low levels in platelets, whereas its paralogue lamellipodin, which can recruit and activate talin in endothelial cells, is not expressed.4 Intriguingly, very recent studies now show that RIAM or alternative Rap1 effectors may be completely dispensable in platelets, because Rap1 can also bind directly to talin.39,40 These interactions are required for integrin activation in vivo, because mutations that prevent them induce integrin activation defects in platelets and, surprisingly, in neutrophils, but not in macrophages.39,40 Although these data explain why αIIbβ3 can still be activated in the absence of RIAM, it remains to be determined why Rap1 needs RIAM to activate β2 integrins. It has been suggested that different integrins localize to distinct membrane compartments that are targeted by distinct signaling pathways.38
Suppression of integrin activation
Dynamic interactions of leukocytes with other cells or matrices require that a proper balance is maintained between active and inactive integrins. Indeed, mice expressing constitutively active mutant α4β1, α4β7, or β2 integrins have strongly augmented leukocyte adhesion to endothelium, which prevents their transmigration.18,41,42 Interestingly, hyperadhesive leukocytes have also been described in an individual with leukocyte defects, whereas platelet function was normal.43 Because mutations in genes encoding leukocyte integrins were not disclosed, this patient may carry a mutation in a protein that suppresses integrin activation.43 Although the importance of reversing leukocyte integrins to their inactive conformation has been long recognized, the underlying mechanisms have remained enigmatic. Potential mechanisms include proteolytic cleavage of integrins and their activators by calpain or phosphorylation of integrin cytoplasmic tails, particularly at the T/S region and the NxxY motifs (Figure 2). It should be noted that mutant mice expressing β3 integrins with Y>F mutations in both motifs develop a mild bleeding phenotype due to platelet defects, whereas the same mutations in β1 do not induce spontaneous abnormalities.17,44 In the following section, we will focus on the suppression of integrin function by negative regulators of integrins.
Cytoplasmic suppressors of integrin activation
Cytoplasmic proteins can prevent integrin activation by 2 scenarios: through indirect effects on other integrin regulators (eg, Rap1) or by direct interactions with integrin cytoplasmic tails. Rap1 activity is prevented by a number of proteins, including Cdc42, RhoH, and the E3 ubiquitin ligase Cbl-b.4 More recent data add Rap1-GTPase activating protein 3 RASA3 to this list (Figure 4A), which inactivates Rap1 by GTP hydrolysis and is abundantly expressed in platelets and megakaryocytes. RASA3 loss of function in mice causes thrombocytopenia as a result of accelerated platelet turnover and leads to increased integrin activation on platelets, which is reversed by deletion of CalDAG-GEF1.45,46 In contrast, no defects are noted in leukocytes. Thus, CalDAG-GEF1 and RASA3 constitute a GEF and GTPase activating protein (GAP) pair for Rap1 that is required for platelet, but not leukocyte, integrin function.
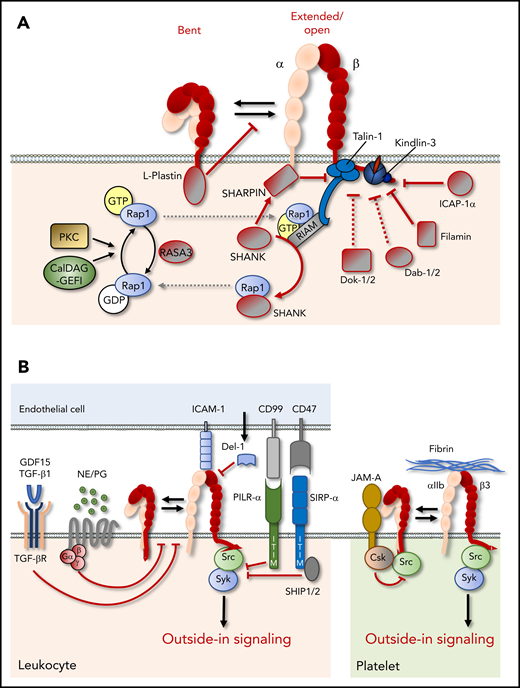
Suppression of integrin function. (A) Cytoplasmic proteins can negatively regulate integrin function through stabilization of the bent integrin conformation, sequestration, or inactivation of integrin activators, such as Rap1, or direct binding to integrin cytoplasmic tails, which may hinder the binding of integrin activators, such as talin and kindlin. (B) Extracellular and cell surface proteins can negatively affect integrin function by preventing inside-out or outside-in signaling. CSK, C-terminal Src kinase; GDF, growth/differentiation factor; ITIM, immunoreceptor tyrosine-based inhibitory motif; JAM-A, junctional adhesion molecule-A; NE, norepinephrine; PG, prostaglandin; SHIP, SH2 domain-containing inositol 5-phosphatase; SIRPα, signal regulatory protein α; TGF-βR, TGF-β receptor.
Suppression of integrin function. (A) Cytoplasmic proteins can negatively regulate integrin function through stabilization of the bent integrin conformation, sequestration, or inactivation of integrin activators, such as Rap1, or direct binding to integrin cytoplasmic tails, which may hinder the binding of integrin activators, such as talin and kindlin. (B) Extracellular and cell surface proteins can negatively affect integrin function by preventing inside-out or outside-in signaling. CSK, C-terminal Src kinase; GDF, growth/differentiation factor; ITIM, immunoreceptor tyrosine-based inhibitory motif; JAM-A, junctional adhesion molecule-A; NE, norepinephrine; PG, prostaglandin; SHIP, SH2 domain-containing inositol 5-phosphatase; SIRPα, signal regulatory protein α; TGF-βR, TGF-β receptor.
Other cytoplasmic proteins can interact directly with integrin tails and may stabilize the clasp or occupy the binding sites for talin and/or kindlin, thereby increasing the threshold for integrin activation. The actin-bundling protein L-plastin, an important regulator of innate and adaptive immunity, binds to clasped αMβ2 and maintains the inactive conformation (Figure 4A), thus preventing leukocyte adhesion to fibrinogen or intercellular adhesion molecule-1 (ICAM-1) under flow.47 Furthermore, several proteins can bind to the membrane-proximal or membrane-distal NxxY motif and, thus, compete with talin or kindlin binding, respectively (Figure 2).48 Although a number of these proteins, such as Numb, the disabled (Dab) proteins, and sorting nexins, regulate integrin endocytosis and recycling, which affects integrin function in a broader sense, others can inhibit integrin inside-out activation, at least in vitro.49,50 Nevertheless, many of the identified proteins seem to bind specifically to some, but not all, β-subunits, and their relevance for the regulation of leukocyte/platelet integrins in vivo is not always clear. For example, integrin cytoplasmic domain-associated protein-1α (ICAP-1α) specifically binds the membrane-distal NxxY motif in β1 (but not in β3 or β5) tails, whereas Dab-2 interacts with the same motif in β3 and β5, but not in β1 or β2, tails (Figures 2 and 4A). Dab-2 is an endocytic adaptor that regulates the internalization of integrins, as well as a wide range of other cell surface proteins; in vivo evidence for its specific role as an inhibitor of integrin activation is lacking.51 The targeted deletion of ICAP-1α in mice results in defects in bone development, putatively due to enhanced osteoblast adhesion and defects in proliferation.52 However, the absence of bleeding complications or an immunological phenotype suggests that ICAP-1α is not crucial for the regulation of integrins on leukocytes or platelets.
In contrast to ICAP-1α and Dab-2, docking protein-1 (Dok-1) and Dok-2, which are highly expressed in immune cells and platelets, bind to the membrane-proximal NxxY motif in a number of β-tails and, thus, compete with talin (Figures 2 and 4A).50 Knockout mouse models support a role for Dok-1 and Dok-2 in the suppression of platelet activation and thrombus formation.53,54 Furthermore, Dok-1/-2 double-knockout mice develop multiple immunological defects, such as lupus-like renal disease and severe experimental colitis.55 Thus, although Dok-1/-2 proteins are clearly involved in the suppression of platelet and immune cell functions, it remains to be determined to what extent these phenotypes reflect integrin-dependent and -independent effects.
Filamins also bind to several integrin β-subunits, including β1, β2, β3, and β7, at a binding site in between the NxxY motifs, partially overlapping with that of talins (Figures 2 and 4A).56 Therefore, filamins compete with talins and possibly also kindlins for integrin binding and can consequently inhibit integrin activation. Nevertheless, filamins are widely expressed proteins that cross-link cytoskeletal filaments and scaffold several cell surface receptors, as well as GTPases, and their GEFs and GAPs. As a result, the defects observed in filamin-knockout mice or in patients carrying a mutation in 1 of the filamin-encoding genes are often hard to interpret, and multiple defects are noted that could alternately result from reduced or increased integrin activation or have no relation to integrins at all.57-59 Thus, although several proteins can occupy the binding sites for talins and kindlins and act as inhibitors of integrin activation in cells, their role in the suppression of leukocyte/platelet integrins in vivo is unclear or remains to be determined.
Intriguingly, the cytoplasmic protein SH3- and multiple ankyrin-repeat domains (SHANK)-associated RH domain interactor (SHARPIN) has been shown to bind the conserved WKxGFFKR sequence in the integrin α1, α2, αL, and α5 subunits (Figures 2 and 4A).60 SHARPIN prevents talin and kindlin interactions with β-tails, thus inhibiting inside-out activation of several β1 integrins and αLβ2.60 Interestingly, it was suggested very recently that SHARPIN can also bind to the β1 cytotail, as well as to kindlin, and that these interactions prevent the interaction of the talin head domain with β1.61 In line with an inhibitory function of SHARPIN on integrins, SHARPIN-deficient leukocytes are strongly adherent and fail to migrate efficiently.62 SHARPIN also binds to the SHANK proteins.63 These are very important in neurons, but their functions in other cell types are still unknown. SHANK proteins contain a domain that is highly homologous to the Rap1-binding region in talin and can bind GTP-bound active Rap1, thus sequestering it away from integrins and precluding integrin activation (Figure 4A).63 Thus, SHARPIN may suppress integrin activation by preventing talin and/or kindlin interactions with integrins, as well as by limiting the amount of active Rap1 at the plasma membrane through SHANK proteins. Because immunological complications in SHANK-deficient mice have not (yet) been described, it is to unclear whether the SHANK proteins play a role in leukocytes. In contrast, SHARPIN-deficient mice develop a chronic multiorgan inflammatory phenotype.64 The lack of regulatory T cells (Tregs), which suppress T cell–dependent autoimmunity, in these mice suggests that SHARPIN plays a role in Treg homeostasis, but the mechanism remains unknown.65 In addition, SHARPIN is a component of the linear ubiquitin chain assembly complex (LUBAC), which promotes NF-κB activation.66 Although the phenotype of SHARPIN-deficient mice is not recapitulated by the deletion of other components of LUBAC, it is possible that the phenotype reflects the combined results of defects in integrin suppression and other SHARPIN-dependent events related to Treg formation or LUBAC function.
Cell surface and extracellular suppressors of integrin activation
In addition to the cytoplasmic regulators discussed in the previous section, integrin function is also strongly regulated by indirect or direct (lateral) interactions with a variety of other transmembrane proteins. Important negative regulators of immune cell and platelet responses, in general, are receptors containing an immunoreceptor tyrosine-based inhibitory motif (ITIM) in their cytoplasmic tails. When phosphorylated, ITIMs sequester phosphatases that inhibit downstream signaling responses by dephosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in their vicinity, thus preventing the docking of Syk and Src-family kinases.67 Although integrins themselves do not contain classical ITAMs, integrin outside-in signaling is strongly associated with Src/Syk-dependent phosphorylation of neighboring receptors (Figure 4B).68,69 For instance, phosphorylation of the ITAMs in FcγRIIA and GPVI-FcRγ promotes integrin αIIbβ3 outside-in signaling in platelets and stimulates thrombus formation.69,70 Consequently, a number of ITIM-containing receptors have been described that can inhibit integrin function, including the platelet receptor PECAM-1 and paired immunoglobulin-like type 2 receptor-α (PILR-α), which is strongly expressed on neutrophils.71,72 PILR-α is a lectin-like molecule that binds to sialic acid–containing O-glycosylated carbohydrates, on the same membrane (in cis) or on the surface of other cells (in trans). PILR-α interactions with ligands in cis negatively regulate β2 integrin–mediated neutrophil adhesion to ICAM-1 and transmigration.72 Whether PILR-α is ligated in cis or in trans appears critical for its function, because it was recently found that neutrophil PILR-α binding to CD99 on the endothelial cell surface (Figure 4B) reinforces neutrophil adhesion under flow.73 Consequently, the absence of CD99 impairs leukocyte attachment to the luminal surface of the endothelium, and causes neutrophil accumulation between venular endothelial cells and the basement membrane in the inflamed cremaster.73
Another ITIM-containing inhibitory receptor that negatively regulates integrin function is SIRPα, which is expressed mainly by myeloid cells and binds to CD47, a marker of “self” abundantly expressed on the surface of most cells (Figure 4B). SIRPα-CD47 interactions provide a “don’t eat me” signal to macrophages and dendritic cells (DCs), which has gained considerable interest in recent years because blockade of SIRPα-CD47 improves the elimination of some tumors, suggesting this could be exploited as an anticancer therapy.74 Several recent studies suggest that the SIRPα-CD47 axis antagonizes a range of functions dependent on integrins αMβ2 and αXβ2, which recognize complement factor iC3b and induce phagocytosis of complement-coated cells and pathogens. First, blockade of SIRPα-CD47 interactions improved phagocytosis of hematopoietic tumor cells by macrophages stimulated by signaling lymphocytic activation molecule F7 in concert with integrin αMβ2.75,76 Second, CD47 deficiency triggered αXβ2/talin-1–dependent target cell capture by DCs and subsequent DC activation, as well as T-cell proliferation and differentiation induced by CD47-deficient cell associated antigen.77 Third, blockade of SIRPα-CD47 interactions improved antibody-mediated cellular cytotoxicity of neutrophils toward tumor cells opsonized with therapeutic antibodies, via a mechanism dependent on integrin αMβ2, Syk, kindlin-3, and actomyosin-based contractility.78
In addition to ITIM-containing receptors, the transmembrane protein junctional adhesion molecule-A (JAM-A) downregulates integrin function. JAM-A associates with αIIbβ3 in resting platelets and recruits C-terminal Src kinase, which suppresses c-Src activation and, thus, precludes the initiation of outside-in signaling. Upon αIIbβ3 activation, JAM-A dissociates from the integrin to permit full outside-in signaling through c-Src.79
Accumulating data indicate that soluble factors can also inhibit integrin function. For example, developmental endothelial locus-1, an ECM protein that is secreted by endothelial cells at the luminal surface, competes with ICAM-1 for binding to integrin αLβ2 on neutrophils (Figure 4B), preventing neutrophil arrest and transmigration.80 Furthermore, 2 recent studies report that cytokines of the transforming growth factor-β superfamily, including growth differentiation factor-15 and transforming growth factor-β1, can inhibit inside-out activation of β2 integrins and α4β1 (Figure 4B) and are required to prevent excessive leukocyte recruitment to inflamed tissues or after myocardial infarction.81,82 Very recently, it has also become apparent that a number of agonists for Gαs-coupled receptors, including prostaglandins, catecholamines, and epinephrine, inhibit β2 integrin activation (Figure 4B) induced by TCR stimulation with antigenic peptides from Epstein-Barr virus or cytomegalovirus.83 The levels of these agonists are generally low during nocturnal sleep, suggesting that circadian rhythms may influence integrin activation in stimulated T cells. Intriguingly, T cells derived from sleep-deprived individuals were much less responsive to TCR-induced integrin activation than were those from individuals who had a normal night’s sleep.83 Although the underlying mechanisms are elusive, these observations could explain the immune-enhancing effects of sleep on a hitherto unrecognized level and open up a new avenue of immunosuppressive strategies targeting integrins.
From molecular mechanisms to clinical solutions
In recent years, a large body of structural, biochemical, cell-biological, and animal studies have provided novel insights into the regulation of hematopoietic integrins and greatly extended our in-depth understanding of integrin behavior in health and disease. The identification of novel integrin regulators will extend diagnostic possibilities by providing novel candidate genes to screen for mutations (eg, in patients who have immunodeficiency due to impaired or exaggerated leukocyte adhesion or who have bleeding complications or an increased risk for thrombosis). In addition, these advances will aid the development of novel therapeutics, such as small molecules, peptides, and antibodies. Currently, a number of hematopoietic integrins are being targeted in the clinic by strategies that interfere with ligand binding. These include integrin αIIbβ3 to prevent thrombosis after percutaneous coronary intervention (abciximab, eptifibatide, and tirofiban), as well as α4 integrins to reduce leukocyte recruitment to the gut in ulcerative colitis and Crohn’s disease (natalizumab and vedolizumab) or the central nervous system in multiple sclerosis (natalizumab).84,85 Although some of these therapeutics are effective and safe, natalizumab can have serious adverse effects, such as an increased risk for progressive multifocal leukoencephalopathy. Furthermore, many other integrin antagonists have had limited effect in clinical trials.84,85 These observations emphasize the ongoing need to further investigate integrin regulation and develop novel classes of antagonists and agonists that more selectively affect certain aspects of integrin function with high specificity, while leaving others intact. The increasingly emerging evidence that individual integrins can behave very differently in terms of conformational rearrangements and the mechanisms that regulate these opens new therapeutic opportunities in this respect. For instance, it was recently shown in preclinical models that the selective blockade of αMβ2 interactions with CD40 ligand reduces leukocyte recruitment, without side effects resulting from the loss of other integrin functions, whereas the partial activation of this integrin protects from end-organ failure associated with lupus.86,87 In addition to therapeutics that directly target integrins, the novel insights into integrin regulation provide tools to promote or suppress integrin function by targeting upstream activators or suppressors. Because it is now becoming clear that individual integrins are controlled by specific subsets of regulators, this knowledge will facilitate future strategies to selectively manipulate integrin function.
Conclusions and perspectives
Recent years have seen a vast increase in our knowledge of hematopoietic integrin regulation, and a plethora of novel regulators has emerged. It is now clear that there are integrin-specific and cell type–specific differences in activation and signaling pathways and that several proteins can suppress integrins in a number of ways. Together, these mechanisms ensure qualitative and quantitative fine-tuning of cell-cell and cell-matrix interactions that are crucial for hemostasis and immunity and offer novel therapeutic strategies to modulate integrin function in disease.
Acknowledgments
The authors thank Arnoud Sonnenberg (The Netherlands Cancer Institute, Amsterdam, The Netherlands) and Monika Wolkers (Sanquin Research) for critical reading of the manuscript. The authors apologize to all colleagues whose work could not be cited because of space constraints. The figures were prepared using templates from Servier Medical Art (https://smart.servier.com).
Work in C.M.’s laboratory is supported by research grants from The Netherlands Organisation for Scientific Research (ZonMW Veni 016.146.160) and the Dutch Thrombosis Foundation (2017-01).
Authorship
Contribution: M.A.N. and C.M. wrote the manuscript and created the figures.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Coert Margadant, Sanquin Research, Plesmanlaan 125, 1066 CX Amsterdam, The Netherlands; e-mail: c.margadant@sanquin.nl.
