

Key Points
Sequential infusion of CAR19/22 T cell is highly active and well tolerated in patients with refractory/relapsed B-cell malignancies.
Dual-targeting of CD19 and CD22 may represent a feasible solution to reduce antigen-escape relapse after CD19/CD22-directed therapies.

Abstract
Antigen-escape relapse has emerged as a major challenge for long-term disease control after CD19-directed therapies, to which dual-targeting of CD19 and CD22 has been proposed as a potential solution. From March 2016 through January 2018, we conducted a pilot study in 89 patients who had refractory/relapsed B-cell malignancies, to evaluate the efficacy and safety of sequential infusion of anti-CD19 and anti-CD22, a cocktail of 2 single-specific, third-generation chimeric antigen receptor-engineered (CAR19/22) T cells. Among the 51 patients with acute lymphoblastic leukemia, the minimal residual disease-negative response rate was 96.0% (95% confidence interval [CI], 86.3-99.5). With a median follow-up of 16.7 months (range, 1.3-33.3), the median progression-free survival (PFS) was 13.6 months (95% CI, 6.5 to not reached [NR]), and the median overall survival (OS) was 31.0 months (95% CI, 10.6-NR). Among the 38 patients with non-Hodgkin lymphoma, the overall response rate was 72.2% (95% CI, 54.8-85.8), with a complete response rate of 50.0% (95% CI, 32.9-67.1). With a median follow-up of 14.4 months (range, 0.4-27.4), the median PFS was 9.9 months (95% CI, 3.3-NR), and the median OS was 18.0 months (95% CI, 6.1-NR). Antigen-loss relapse occurred in 1 patient during follow-up. High-grade cytokine release syndrome and neurotoxicity occurred in 22.4% and 1.12% patients, respectively. In all except 1, these effects were reversible. Our results indicated that sequential infusion of CAR19/22 T cell was safe and efficacious and may have reduced the rate of antigen-escape relapse in B-cell malignancies. This trial was registered at www.chictr.org.cn as #ChiCTR-OPN-16008526.
Introduction
Immunotherapy is one of the most promising fields of cancer research and treatment.1-5 Clinical trials of second-generation, CD19-targeted, chimeric antigen receptor–engineered (CAR19) T-cell therapy have elicited unprecedented responses.6-11 The therapy achieves complete response (CR) in 90% of patients with refractory/relapsed (r/r) B-cell acute lymphoblastic leukemia (B-ALL) and 50% of patients with r/r B-cell non-Hodgkin lymphoma (B-NHL).6-11 However, CD19− relapse represents one of the most frequent causes of treatment failure and confers dismal outcomes in patients after CD19-directed therapies, an effect that has been observed in ∼10% to 30% of enrolled patients across clinical trials and has accounted for 73.7% of relapses in phase 2 of the Efficacy and Safety of CTL019 in Pediatric Patients With Relapsed and Refractory B-cell Acute Lymphoblastic Leukemia Trial.12-15 Driven by potent selective pressures under CAR19 T cells or blinatumomab, multiple antigen-escape tactics, such as loss of recognizable CD19 epitope because of mutation, alternative splicing, or protein misfolding of CD1916-19 and myeloid lineage switch of CD19+ malignant cells,20,21 have been developed by malignant B cells.
Novel therapeutic strategies have been actively explored preclinically or in clinical trials to overcome CD19− relapse, including transplantation or combinational antigen targeting of multiple tumor-associated antigens.12-14 Administration of 2 single-specific CAR T cells against CD19 and CD123 was shown to efficiently eradicate CD19− cells and prevent antigen-escape relapses in a xenograft model.22 Recently, the CD22-targeted CAR (CAR22) T cell was revealed to induce durable remissions in patients who were resistant to prior CD19-directed immunotherapies,23 given that CD22 usually retains its expression on the surface of CD19− leukemia cells.23,24 Intriguingly, diminished site density, rather than mutation or alternative splicing of CD22, was ascribed to relapses after CAR22 therapy.23 All of these fueled our interests in dual CD19 and CD22 targeting to overcome antigen-escape relapse after CD19- or CD22-directed therapies. However, its potency in preventing antigen loss has not yet been tested, and its efficacy and safety should be evaluated.
We conducted an open-label, single-center, single-arm pilot study to explore the efficacy and safety of sequential infusion of a cocktail of anti-CD19 and -CD22, 2 single-specific, third-generation CAR (CAR19/22) T cells in patients with r/r B-ALL/NHL.
Methods
Study design and procedures
This study design was approved by the institutional review board of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. Written informed consent was obtained from each participant, in compliance with the Declaration of Helsinki.
Eligible patients had relapsed or were refractory to their previous treatments, including autologous or allogenic hematopoietic stem cell transplantation (auto-/allo-HSCT). They were diagnosed according to the World Health Organization classification for tumors of the hematopoietic and lymphoid tissues. Dual expression of CD19 and CD22 on malignant B cells was confirmed by flow cytometry or immunohistochemistry. Good performance status (Eastern Cooperative Oncology Group-Performance Status ≤2), essentially normal organ function, measurable disease, and a life expectancy of 12 weeks or more were necessary for eligibility, whereas patients with uncontrollable infection, active graft-versus-host disease, or clinically evident neurological lesions were excluded (supplemental Methods, available on the Blood Web site).
Patients were given fludarabine 25 mg/m2 and cyclophosphamide 300 mg/m2 for 3 days (days −4 to −2) for lymphodepletion chemotherapy. CAR19 and CAR22 T cells were infused separately on successive days from day 0 (supplemental Figure 1). Bone marrow aspiration or diagnostic imaging was performed for response assessment every month for half a year and every 3 months thereafter. All patients were followed up until they died, were lost to follow-up, or withdrew consent. Cytokine release syndrome (CRS) was graded as the scale proposed by Lee et al.25 CAR T-cell–related encephalopathy syndrome (CRES) and other adverse events (AEs) were evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events v.4.03.26,27 Staging and response assessments were defined according to the National Comprehensive Cancer Network guidelines and Lugano Treatment Response Criteria.28 Further details regarding the study procedures are described in the supplemental Methods.
Lentiviral construction and cell production
The third-generation CAR used in this trial was composed of a single-chain variable fragment derived from a murine monoclonal antibody against human CD19 or CD22, 2 costimulatory domains from CD28 and 4-1BB, and the CD3-ζ chain as the activation domain (supplemental Figure 2). Validation of the CAR constructs and procedures for cell production and quality control assays are described in the supplemental Methods.29,30
Laboratory assessments
Multiparameter flow cytometry (MFC) was used to screen and quantitate minimal residual disease (MRD) in blood, bone marrow (BM), and cerebrospinal fluid. In vivo expansion of CAR19 and CAR22 T cells were measured by droplet digital polymerase chain reaction. Cytokines were assessed according to the manufacturer’s instructions. Cytogenetic and genomic aberrations were identified by karyotyping, real-time quantitative polymerase chain reaction, fluorescence in situ hybridization, and next-generation exome sequencing. Detailed methods have been previously described.31,32
Statistical analysis
Progression-free survival (PFS) and overall survival (OS) were estimated as the time from first infusion to first relapse or death, respectively. The analysis of categorical variables was performed using the Clopper-Pearson 95% confidence interval (CI) and Fisher’s exact test. Wilcoxon rank-sum test was applied to continuous variables. Cumulative incidence of relapse (CIR) were calculated according to the Fine and Gray method, with nonrelapse mortality and subsequent allo-HSCT considered as competing risks.33 Gray’s method was used to evaluate the differences between groups. The probabilities of OS and PFS were estimated by means of the Kaplan-Meier method and were compared with the use of the log-rank test. Analyses were performed with the use of GraphPad Prism 7 and Statistical Analysis Software. Values of P < .05 (2-tailed) were considered statistically significant.
Results
Baseline characteristics
From March 2016 through January 2018, 105 patients with r/r B-ALL/NHL were screened for eligibility in this study. Because of production failure, serious infection, withdrawal of consent, disease progression, and death, among other factors, 16 patients who did not receive qualified CAR19/22 T-cell cocktail infusion were excluded from the study analysis (supplemental Figure 3). A total of 89 patients (84.8%) finished the CAR19/22 T-cell cocktail infusion and were included in the analysis, including 51 B-ALL patients and 38 B-NHL patients. The cutoff date for data collection was 31 March 2019.
The baseline characteristics of these patients are summarized in supplemental Table 1. The median age was 36 years (range, 9-71) with 87.6% aged ≥18 years. In the B-ALL cohort, the median percentage of BM blasts was 59.0% (range, 0.5%-97.5%). High-risk cytogenetic and genomic aberrations were detected in 36 patients (70.6%; supplemental Table 1). In the B-NHL cohort, all patients manifested aggressive clinical courses. Twenty-three patients (60.5%) relapsed ≥3 times or had primary refractory diseases. Chromosome 17p deletion (del[17p]), TP53 mutation, and IgH/MYC translocation were detected in 17 patients (44.7%), including 4 concurrently carrying MYC and BCL2 or BCL6 translocations (double-hit high-grade B-cell lymphoma [HGBL DH]; supplemental Table 1).
Manufacturing and infusion of CAR22 and CAR19 T-cells
Functional validation of the CAR constructs is presented in supplemental Figures 4-6, which show the results of transduction efficiency, apoptosis (supplemental Figure 4), the antileukemic effects of CAR19 and CAR22 T cell in vitro (supplemental Figure 5) and in xenograft mice models (supplemental Figure 6).
The median manufacturing time was 12 days for CAR19 T cells (range, 8-20 days) and 13 days for CAR22 (range, 9-22 days) T cells. Mean transduction efficiency of CAR19 and CAR22 T cells were 40.4% ± 18.4% and 42.8% ± 19.6%, respectively. In general, CAR19 and CAR22 T cells were given separately in 2 divided doses (supplemental Table 2) and infused on successive days from day 0 (supplemental Figure 1). B-ALL patients received 2.6 ± 1.5×106/kg CAR19 T cells and 2.7 ± 1.2×106/kg CAR22 T cells; B-NHL patients received higher doses (ie, 5.1 ± 2.1 ×106/kg CAR19 T cells and 5.3 ± 2.4 ×106/kg CAR22 T cells). Eighty-one patients (91.0%) received CAR22 T cells first. The expansion of CAR19 and CAR22 T cells was validated in the peripheral blood from all patients (Figure 1; supplemental Table 3).
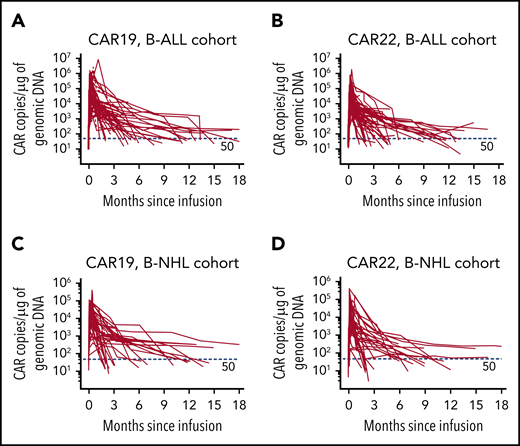
Cellular kinetics of CAR19 and CAR22 transgenes in peripheral blood. Copies of CAR19 (A) and CAR22 (B) transgenes, respectively, in patients with B-ALL and copies of CAR19 (C) and CAR22 (D) transgenes in patients with B-NHL. Genomic DNA was isolated from the samples of whole blood at serial time points before and after infusion of the CAR T-cell cocktail. Each panel represents the cellular kinetics of CAR transgenes within the first 18 months since infusion. The red horizontal line in each panel denotes the lower limit of quantitation (50 copies/μg).
Cellular kinetics of CAR19 and CAR22 transgenes in peripheral blood. Copies of CAR19 (A) and CAR22 (B) transgenes, respectively, in patients with B-ALL and copies of CAR19 (C) and CAR22 (D) transgenes in patients with B-NHL. Genomic DNA was isolated from the samples of whole blood at serial time points before and after infusion of the CAR T-cell cocktail. Each panel represents the cellular kinetics of CAR transgenes within the first 18 months since infusion. The red horizontal line in each panel denotes the lower limit of quantitation (50 copies/μg).
Outcomes in patients with B-ALL
At the day 30 assessment, MRD− CR (<0.01% BM blasts) or CR with incomplete count recovery (CRi), determined by MFC assays, was achieved in 48 B-ALL patients (96.0%; 95% CI, 86.3-99.5), including 12 patients (100%; 95% CI, 73.5-100) with prior transplantation, 13 patients (100%; 95% CI, 75.3-100) with Ph chromosome (Ph+), 11 patients (100%; 95% CI, 71.5-100) with del(17p) or TP53 mutations, 4 patients (100%; 95% CI, 39.8-100) with hypodiploidy, 5 of the 6 patients (83.3%; 95% CI, 35.9-99.6) with MLL rearrangements (MLLr, 4 with MLL-AF4), and 3 of the 4 patients (75.0%; 95% CI, 19.4-99.4) with Ph-like ALL. One patient with MLL-AF4 rearrangement and 1 patient with the JAK2 R683G mutation (Ph-like ALL) had partial response (PR) and stable disease, respectively, and progressed rapidly within 2 months. The remaining patient, who died of serious infection and CRS within the first month, was excluded from the response evaluation but was included in the toxicity and survival assessments (supplemental Figure 3). The overall response rate (ORR; CR+CRi) was similar across subgroups with different clinical features (supplemental Table 4).
Of the 49 patients who had a response, 24 (49.0%, 24/49) patients thereafter had a relapse. The CIR at month 12 was 0.291 (95% CI, 0.200-0.423; Figure 2A). Except for patients with central nervous system (CNS) involvement (CNS 2; P = .033; Figure 2B), CIR was consistent in different subgroups (supplemental Figure 7). Notably, of the 24 patients, 23 relapsed with CD19/CD22–double-positive disease. Expression of CD19 and CD22 was identified on the surface of lymphoblasts from these patients, with CAR19 and/or CAR 22 transgenes detected in 8 patients and B-cell aplasia observed in 5 patients at the time of relapse. One patient with TP53 mutation had a CD19−/CD22dim relapse at 9.3 months after CAR T-cell infusion, with CAR19, CAR 22 transgenes, and B-cell aplasia sustained.
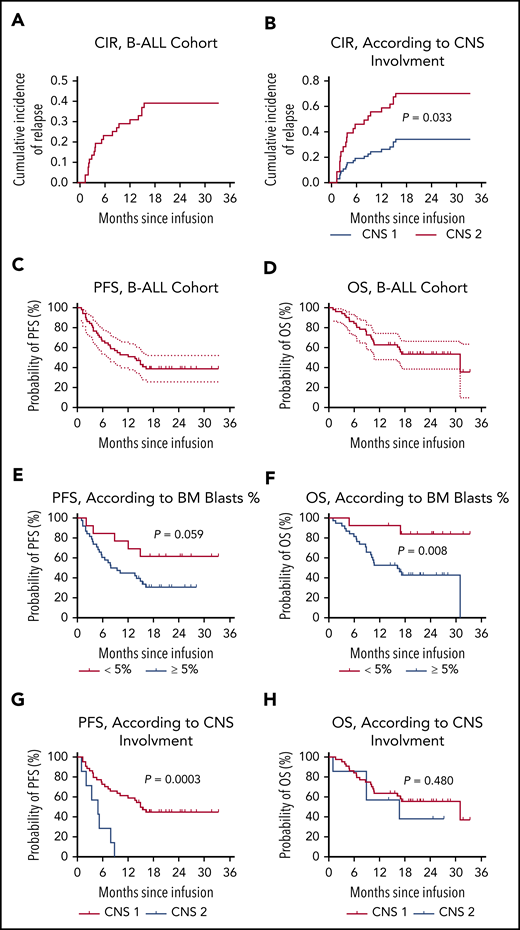
Outcomes in patients with B-ALL after CAR19/22 T-cell cocktail therapy. (A) CIR in B-ALL cohort. CIR was calculated, with nonrelapse mortality and subsequent allo-HSCT considered as competing risks. Patients who did not have a relapse at last follow-up were censored. The CIR at month 12 was 0.291 (95% CI, 0.200-0.423). (B) CIR according to CNS involvement at enrollment. Relapse was more frequent in patients with CNS involvement (CNS 2) compared with patients without (CNS 1; P = .033). PFS (C) and OS (D) in this cohort. The median PFS was 13.6 months (95% CI, 6.5-NR), and the median OS was 31.0 months (95% CI, 10.6-NR). PFS (E) and OS (F) according to leukemia burden in BM. In the patients with low leukemia burden (<5% BM blasts) at baseline, the median PFS and OS were NR. In the patients with high leukemia burden (≥5% BM blasts) at baseline, the median PFS was 8.6 months (95% CI, 5.2-15.4) and the median OS was 16.2 months (95% CI, 8.9-31.0). PFS (G) and OS (F), according to CNS involvement. In the patients without CNS involvement (CNS 1) at baseline, the median PFS was 14.9 months (95% CI, 7.8-NR), and the median OS was 31.0 months (95% CI, 10.6-NR). In the patients with CNS involvement (CNS 2) at baseline, the median PFS was 4.9 months (95% CI, 1.0-7.9), and the median OS was 16.7 months (95% CI, 1.0-NR).
Outcomes in patients with B-ALL after CAR19/22 T-cell cocktail therapy. (A) CIR in B-ALL cohort. CIR was calculated, with nonrelapse mortality and subsequent allo-HSCT considered as competing risks. Patients who did not have a relapse at last follow-up were censored. The CIR at month 12 was 0.291 (95% CI, 0.200-0.423). (B) CIR according to CNS involvement at enrollment. Relapse was more frequent in patients with CNS involvement (CNS 2) compared with patients without (CNS 1; P = .033). PFS (C) and OS (D) in this cohort. The median PFS was 13.6 months (95% CI, 6.5-NR), and the median OS was 31.0 months (95% CI, 10.6-NR). PFS (E) and OS (F) according to leukemia burden in BM. In the patients with low leukemia burden (<5% BM blasts) at baseline, the median PFS and OS were NR. In the patients with high leukemia burden (≥5% BM blasts) at baseline, the median PFS was 8.6 months (95% CI, 5.2-15.4) and the median OS was 16.2 months (95% CI, 8.9-31.0). PFS (G) and OS (F), according to CNS involvement. In the patients without CNS involvement (CNS 1) at baseline, the median PFS was 14.9 months (95% CI, 7.8-NR), and the median OS was 31.0 months (95% CI, 10.6-NR). In the patients with CNS involvement (CNS 2) at baseline, the median PFS was 4.9 months (95% CI, 1.0-7.9), and the median OS was 16.7 months (95% CI, 1.0-NR).
Persistent CAR transgenes were observed in the peripheral blood of B-ALL patients, with median times of ∼10 months (Figure 1A-B; supplemental Table 3). The median times for the emergence of BM hematogones and for the recovery of peripheral B cells were 4.0 months (range, 1.0-18.4) and 8.8 months (range, 0.5-18.4), respectively (supplemental Figure 8; supplemental Table 3). The median PFS of the entire cohort was 13.6 months (95% CI, 6.5-not reached [NR]; Figure 2C), and the median OS was 31.0 months (95% CI, 10.6-NR; Figure 2D), with a median follow-up of 16.7 months (range, 1.3-33.3). The 12-month PFS rate was 52.9% (95% CI, 38.5-65.5) and the 12-month OS rate was 62.8% (95% CI, 48.0-74.4). Patients with lower leukemia burden (< 5% BM blasts) at baseline had better OS (P = .008) than patients with higher leukemia burden (≥5% BM blasts), although a marginal statistical benefit in PFS (P = .059; Figure 2E-F). Likewise, patients with CNS involvement (CNS 2) had marked reduced PFS (P = .0003), compared with patients without involvement (CNS 1; Figure 2G-H). Furthermore, 12 patients proceeded to allo-HSCT (supplemental Figure 9A); however, no survival benefit was achieved with consolidated HSCT (supplemental Figure 9B-C).
Subtype-specific prognostic impact was investigated in the B-ALL cohort. Of the 15 patients without high-risk cytogenetic and genomic aberrations, the 12-month PFS rate was 60.0% (95% CI, 31.8-79.7) and the median PFS was 14.9 months (95% CI, 5.8-NR), with a median follow-up of 16.0 months (range, 4.8-31.7) (Figure 3A). Intriguingly, no significant difference in either PFS or OS was found among patients with or without high-risk genetic changes which included BCR/ABL rearrangement with or without T315I mutation, MLLr, hypodiploidy, del(17p) or TP53 mutations, Ph-like ALL and RAS pathway mutations, although the number of patients in some subgroups was relatively small (Figure 3A-B). Of the 13 patients with Ph+ B-ALL, the median PFS was 15.4 months (95% CI, 3.4-NR) and the 12-month PFS rate was 53.9% (95% CI, 24.8-76.0), with a median follow-up of 25.0 months (range, 8.9-33.3; Figure 3C). Seven patients (3 with T315I mutation) had an MRD+ relapse, determined by MFC or BCR/ABL1 real-time qPCR, and treatments with tyrosine kinase inhibitors (4 with dasatinib and 3 with ponatinib) were initiated. All of them achieved second remissions, and 3 proceeded to allo-HSCT. However, improved OS (P = .028; Figure 3C) but not PFS (P = .556; Figure 3D) was demonstrated in patients with Ph+ B-ALL compared with patients with Ph− B-ALL.

Prognostic impact of genetic abnormalities in patients with B-ALL after CAR19/22 T-cell cocktail therapy. PFS (A) and OS (B), according to risk-stratified genetic subtypes. In the patients with Ph+, the median PFS was 15.4 months (95% CI, 3.4-NR), and the median OS was 31.0 months (95% CI, 31.0-NR). In the patients with hypodiploidy, the median PFS was NR (95% CI, 3.8-NR), and the median OS was 31.0 months (95% CI, 3.8-31.0). In the patients with T315I mutation in ABL kinase, the median PFS and median OS were NR. In the patients with del(17p) or the TP53 mutation, the median PFS was 14.0 months (95% CI, 4.9-NR), and the median OS was NR. In the patients with MLL rearrangements, the median PFS and median OS were NR. In the patients with Ph-like B-ALL, the median PFS was 6.2 months (95% CI, 2.0-NR), and the median OS was 8.5 months (95% CI, 5.9-NR). In the patients with RAS pathway mutations, including mutations in genes NRAS, KRAS, and PTPN11, the median PFS was 2.0 months (95% CI, 1.0-16.2), and the median OS was 4.9 months (95% CI, 1.0-17.2). In the patients without the above-mentioned aberrations (W/O), the median PFS was 14.9 months (95% CI, 5.8-NR), and the median OS was NR (95% CI, 7.3-NR). PFS (C) and OS (D), according to Ph status. In the patients with Ph+, the median PFS was 15.4 months (95% CI, 3.4-NR), and the median OS was 31.0 months (95% CI, 31.0-NR). In the patients with Ph−, the median PFS was 12.8 months (95% CI, 5.8-NR), and the median OS was 16.7 months (95% CI, 8.9-NR).
Prognostic impact of genetic abnormalities in patients with B-ALL after CAR19/22 T-cell cocktail therapy. PFS (A) and OS (B), according to risk-stratified genetic subtypes. In the patients with Ph+, the median PFS was 15.4 months (95% CI, 3.4-NR), and the median OS was 31.0 months (95% CI, 31.0-NR). In the patients with hypodiploidy, the median PFS was NR (95% CI, 3.8-NR), and the median OS was 31.0 months (95% CI, 3.8-31.0). In the patients with T315I mutation in ABL kinase, the median PFS and median OS were NR. In the patients with del(17p) or the TP53 mutation, the median PFS was 14.0 months (95% CI, 4.9-NR), and the median OS was NR. In the patients with MLL rearrangements, the median PFS and median OS were NR. In the patients with Ph-like B-ALL, the median PFS was 6.2 months (95% CI, 2.0-NR), and the median OS was 8.5 months (95% CI, 5.9-NR). In the patients with RAS pathway mutations, including mutations in genes NRAS, KRAS, and PTPN11, the median PFS was 2.0 months (95% CI, 1.0-16.2), and the median OS was 4.9 months (95% CI, 1.0-17.2). In the patients without the above-mentioned aberrations (W/O), the median PFS was 14.9 months (95% CI, 5.8-NR), and the median OS was NR (95% CI, 7.3-NR). PFS (C) and OS (D), according to Ph status. In the patients with Ph+, the median PFS was 15.4 months (95% CI, 3.4-NR), and the median OS was 31.0 months (95% CI, 31.0-NR). In the patients with Ph−, the median PFS was 12.8 months (95% CI, 5.8-NR), and the median OS was 16.7 months (95% CI, 8.9-NR).
Outcomes in B-NHL patients
At a minimum follow-up of 3 months, 26 (72.2%; 95% CI, 54.8-85.8) of the 36 evaluable patients achieved overall responses, including 18 (50%; 95% CI, 32.9-67.1) with a CR and 8 (22.2%; 95% CI, 10.1-39.2) with a PR (Figure 4A). Two additional patients who died of septic shock and severe CRS within the first month were excluded from response evaluation but were included in the toxicity and survival assessments (supplemental Figure 3). The ORR (CR+PR) at month 3 was consistent in different subgroups, irrespective of pathologic subtypes (eg, cell of origin and cytogenetic or genomic aberrations; supplemental Table 5).
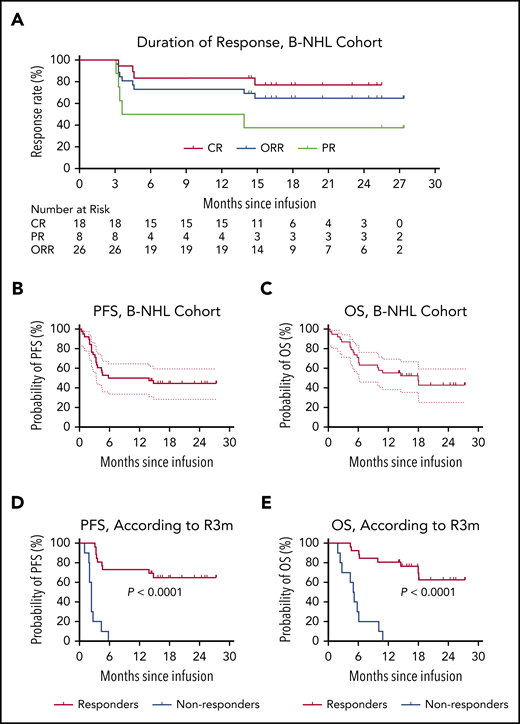
Outcomes in patients with B-NHL after CAR19/22 T-cell cocktail therapy. (A) The duration of response in the 26 patients with B-NHL who achieved an overall response, including patients with CR and those with a PR. PFS (B) and OS (C) in the entire B-NHL cohort. The median PFS was 9.9 months (95% CI, 3.3-NR), and the median OS was 18.0 months (95% CI, 6.1-NR). PFS (D) and OS (E), according to the response status at month 3 after CAR19/22 T-cell cocktail therapy. Of the patients who achieved an overall response at month 3 (responders), the median PFS and median OS were NR. Of the patients who did not have a response at month 3 (nonresponders), the median PFS was 2.4 months (95% CI, 1.0-2.7), and the median OS was 5.1 months (95% CI, 1.9-6.1).
Outcomes in patients with B-NHL after CAR19/22 T-cell cocktail therapy. (A) The duration of response in the 26 patients with B-NHL who achieved an overall response, including patients with CR and those with a PR. PFS (B) and OS (C) in the entire B-NHL cohort. The median PFS was 9.9 months (95% CI, 3.3-NR), and the median OS was 18.0 months (95% CI, 6.1-NR). PFS (D) and OS (E), according to the response status at month 3 after CAR19/22 T-cell cocktail therapy. Of the patients who achieved an overall response at month 3 (responders), the median PFS and median OS were NR. Of the patients who did not have a response at month 3 (nonresponders), the median PFS was 2.4 months (95% CI, 1.0-2.7), and the median OS was 5.1 months (95% CI, 1.9-6.1).
At the data cutoff date, 15 of the 18 patients who had a CR at month 3 maintained their responses, and 3 of 8 patients who had a PR within 3 months continued to have a CR, with a median time of 15.0 months (range, 12.0-17.0) without additional therapy. Of the 11 (28.9%) patients who subsequently underwent transplantation (9 with auto-HSCT and 2 with allo-HSCT), 6 maintained their initial CRs without progressive disease, and 1 with HGBL DH had ongoing CR for 25.0 months at the data cutoff date. Collectively, the best ORR was 83.3% (95% CI, 67.2-93.6), with a best CR rate of 58.3% (95% CI, 40.8-74.5) and a best PR rate of 25.0% (95% CI, 12.1-42.2).
With a median follow-up of 14.4 months (range, 0.4-27.4), the median PFS was 9.9 months (95% CI, 3.3-NR; Figure 4B), and the median OS was 18.0 months (95% CI, 6.1-NR; Figure 4C) in the B-NHL cohort. The 12-month PFS rate was 50.0% (95% CI, 33.4-64.5; Figure 4B) and the 12-month OS rate was 55.3% (95% CI, 38.3-69.3; Figure 4C). Patients who received therapy with the CAR19/22 T-cell cocktail at first relapse had longer survival than those who received therapy for primary refractory disease or multiple relapses (supplemental Figure 10). Notably, patients who achieved an overall response at month 3 (R3m, defined as complete or partial response at month 3 after CAR19/22 T-cell therapy) had significantly extended PFS (P < .0001) and OS (P < .0001) when compared with others (Figure 4D-4E; supplemental Figure 11). In contrast, disease progression eventually occurred in 18 (50.0%) patients, with a median time to progression of 3.3 months (range, 1.0-14.8). At the time of disease progression, CAR19 and/or CAR22 transgenes were detected in 2 patients, and B-cell aplasia was observed in 3 patients. Repeat biopsy and immunophenotyping were conducted in 7 (38.9%) of the 18 patients. However, loss of CD19 or CD22 was not detected. Expression of CD19 and CD22 was revealed by immunohistochemistry or MFC in the neoplastic B cells from these patients.
Subtype-specific prognostic impact was also explored in the B-NHL cohort. Of the 23 patients with diffuse large B-cell lymphoma, not otherwise specified, with a median follow-up of 14.3 months (range, 0.4-27.3), the 12-month PFS rate was 47.8% (95% CI, 26.8-66.1; supplemental Figure 12A). The median PFS was 5.8 months (95% CI, 3.0-NR; supplemental Figure 12A), and the median OS was 18.0 months (95% CI, 5.2-NR; supplemental Figure 12B). The 15 patients (65.2%; 95% CI, 42.7-83.6) with R3m had significantly prolonged PFS (P < .0001; supplemental Figure 12C) and OS (P < .0001; supplemental Figure 12D) compared with those without. Of the 9 patients with IgH/MYC translocation, the median PFS and median OS were NR (Figure 5). With a median follow-up of 16.6 months (range, 2.7-27.3), the 12-month PFS rate was 66.7% (95% CI, 28.2-87.8; Figure 5). At the data cutoff date, 6 (66.7%; 95% CI, 29.9-92.5) of the 7 patients who had achieved R3m maintained their responses, including 3 patients with HGBL DH in CR. However, of the 10 patients with del(17p) or the TP53 mutation, the 12-month PFS rate was 50.0% (95% CI, 18.4-75.3) with a median follow-up of 12.5 months (range, 2.7-25.5). The median PFS was 8.8 months (95% CI, 1.0-NR) and the median OS was 14.5 months (95% CI, 2.7-NR), but there were no significant differences in PFS or OS when compared with other genetic subgroups (Figure 5).
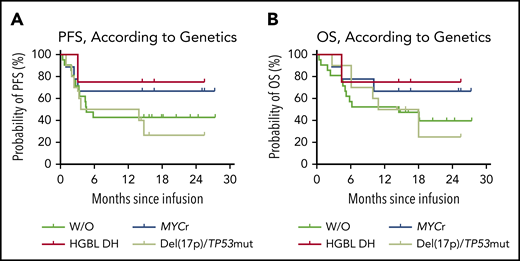
Prognostic impact of genetic abnormalities in patients with B-NHL after CAR19/22 T-cell cocktail therapy. PFS (A) and OS (B), according to risk-stratified genetic subtypes. Of the patients with MYC rearrangements (MYCr), including patients with MYC and BCL2 or BCL6 rearrangements (HGBL DH), the median PFS and median OS were NR. Two patients with HGBL DH further received auto-HSCT and second-round CAR T-cell cocktail infusion. Of the patients with del(17p) or TP53 mutation, the median PFS was 8.8 months (95% CI, 1.0-NR), and the median OS was 14.5 months (95% CI, 2.7-NR). Of the patients without the above-mentioned aberrations (W/O), the median PFS was 4.6 months (95% CI, 3.0-NR), and the median OS was 14.5 months (95% CI, 4.6-NR).
Prognostic impact of genetic abnormalities in patients with B-NHL after CAR19/22 T-cell cocktail therapy. PFS (A) and OS (B), according to risk-stratified genetic subtypes. Of the patients with MYC rearrangements (MYCr), including patients with MYC and BCL2 or BCL6 rearrangements (HGBL DH), the median PFS and median OS were NR. Two patients with HGBL DH further received auto-HSCT and second-round CAR T-cell cocktail infusion. Of the patients with del(17p) or TP53 mutation, the median PFS was 8.8 months (95% CI, 1.0-NR), and the median OS was 14.5 months (95% CI, 2.7-NR). Of the patients without the above-mentioned aberrations (W/O), the median PFS was 4.6 months (95% CI, 3.0-NR), and the median OS was 14.5 months (95% CI, 4.6-NR).
AEs
The most common severe AEs (grade 3 or higher) within the first month were cytopenias (Table 1). A total of 85 (95.5%) patients experienced CRS; 77.6% were of low grade (grade 1-2). Except for 1 patient with B-ALL (grade 5; 1.2%), all high-grade (grade 3 or higher; 22.4%) cases were reversible (supplemental Tables 6-7). CRES developed in 12 (13.5%) patients and was reversible in all of them. Except for 1 patient with B-ALL (grade 4; 1.12%), all the patients with CRES were of low grade (grade 1-2; supplemental Tables 6-7). The incidences of CRS and CRES were similar between the B-ALL and B-NHL cohorts, regardless of severity (P = .953 for the B-ALL cohort; P = .938 for the B-NHL cohort; supplemental Table 8).
AEs occurring in at least 5% of patients within the first month after CAR T-cell infusion
| AE | Grades 1-2, n (%) | Grade 3, n (%) | Grade 4, n (%) | Grade 5, n (%) |
|---|---|---|---|---|
| Lymphocytopenia | 0 | 0 | 89 (100) | 0 |
| Neutrocytopenia | 0 | 2 (2) | 87 (98) | 0 |
| Leukopenia | 1 (1) | 6 (7) | 82 (92) | 0 |
| Anemia | 16 (18) | 64 (72) | 9 (10) | 0 |
| CRS | 66 (74) | 15 (17) | 3 (3) | 1 (1) |
| Fever | 52 (58) | 32 (36) | 0 | 0 |
| Thrombocytopenia | 27 (30) | 10 (11) | 46 (52) | 0 |
| Hypotension | 73 (82) | 8 (9) | 0 | 0 |
| APTT prolonged | 66 (74) | 5 (6) | 0 | 0 |
| Sinus tachycardia | 67 (75) | 0 | 0 | 0 |
| Hypoalbuminemia | 63 (71) | 1 (1) | 0 | 0 |
| Hypoxia | 50 (56) | 2 (2) | 0 | 0 |
| Hypokalemia | 29 (33) | 12 (13) | 3 (3) | 0 |
| ALT increase | 28 (31) | 3 (3) | 0 | 0 |
| Alkaline phosphatase increase | 27 (30) | 1 (1) | 0 | 0 |
| Diarrhea | 20 (22) | 5 (6) | 0 | 0 |
| AST increase | 18 (20) | 6 (7) | 0 | 0 |
| Lung infection | 0 | 16 (18) | 0 | 3 (3) |
| Hypocalcemia | 14 (16) | 3 (3) | 0 | 0 |
| CRES | 11 (12) | 0 | 1 (1) | 0 |
| Fibrinogen decreased | 7 (8) | 4 (4) | 1 (1) | 0 |
| Hyponatremia | 10 (11) | 1 (1) | 0 | 0 |
| Heart failure | 4 (4) | 1 (1) | 0 | 0 |
| AE | Grades 1-2, n (%) | Grade 3, n (%) | Grade 4, n (%) | Grade 5, n (%) |
|---|---|---|---|---|
| Lymphocytopenia | 0 | 0 | 89 (100) | 0 |
| Neutrocytopenia | 0 | 2 (2) | 87 (98) | 0 |
| Leukopenia | 1 (1) | 6 (7) | 82 (92) | 0 |
| Anemia | 16 (18) | 64 (72) | 9 (10) | 0 |
| CRS | 66 (74) | 15 (17) | 3 (3) | 1 (1) |
| Fever | 52 (58) | 32 (36) | 0 | 0 |
| Thrombocytopenia | 27 (30) | 10 (11) | 46 (52) | 0 |
| Hypotension | 73 (82) | 8 (9) | 0 | 0 |
| APTT prolonged | 66 (74) | 5 (6) | 0 | 0 |
| Sinus tachycardia | 67 (75) | 0 | 0 | 0 |
| Hypoalbuminemia | 63 (71) | 1 (1) | 0 | 0 |
| Hypoxia | 50 (56) | 2 (2) | 0 | 0 |
| Hypokalemia | 29 (33) | 12 (13) | 3 (3) | 0 |
| ALT increase | 28 (31) | 3 (3) | 0 | 0 |
| Alkaline phosphatase increase | 27 (30) | 1 (1) | 0 | 0 |
| Diarrhea | 20 (22) | 5 (6) | 0 | 0 |
| AST increase | 18 (20) | 6 (7) | 0 | 0 |
| Lung infection | 0 | 16 (18) | 0 | 3 (3) |
| Hypocalcemia | 14 (16) | 3 (3) | 0 | 0 |
| CRES | 11 (12) | 0 | 1 (1) | 0 |
| Fibrinogen decreased | 7 (8) | 4 (4) | 1 (1) | 0 |
| Hyponatremia | 10 (11) | 1 (1) | 0 | 0 |
| Heart failure | 4 (4) | 1 (1) | 0 | 0 |
APTT, activated partial thromboplastin time; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
In both disease cohorts, patients with high-grade CRS had significantly higher peak concentrations of serum interleukin-6 (IL-6) (P = .002 for B-ALL, Figure 6A; P = .000 for B-NHL, Figure 6B) and serum ferritin (SF; P = .004 for B-ALL, Figure 6C; P = .000 for B-NHL, Figure 6D) than those with low-grade CRS or without CRS. High-grade CRS was also markedly associated with higher percentages of BM blasts (P = .006; Figure 6E) in the B-ALL cohort, and was associated with increased levels of serum C-reactive protein (P = .003; Figure 6F) and higher International Prognostic Index scores (P = .025; Figure 6G) in the B-NHL cohort, but was not associated with disease stage and dosages of CAR T cells in both disease cohorts. A similar association was found between the occurrence of CRES and the increased levels of IL-6 (P = .042; Figure 6H) and SF (P = .034; Figure 6I) in the B-ALL cohort.

Biomarkers correlating with CRS and CRES. The peak concentrations of serum IL-6 (sIL-6) associated with the severity of CRS in the cohort of B-ALL (A) and the cohort of B-NHL (B). The peak concentrations of SF associated with the severity of CRS in the cohort of B-ALL (C) and the cohort of B-NHL (D). (E) The baseline percentage of blasts in BM associated with the severity of CRS in the cohort of B-ALL. (F) The peak concentration of serum C-reactive protein (sCRP) associated with the severity of CRS in the cohort of B-NHL. (G) The international prognostic index (IPI) score at baseline associated with the severity of CRS in the cohort of B-NHL. The peak concentrations of sIL-6 (H) and SF (I), associated with the occurrence of CRES in the cohort of B-ALL. The horizontal lines in each box represent the median values, and the lower and upper borders of each box represent the 25th and the 75th percentiles, respectively, and the whiskers represent the minimum and maximum range.
Biomarkers correlating with CRS and CRES. The peak concentrations of serum IL-6 (sIL-6) associated with the severity of CRS in the cohort of B-ALL (A) and the cohort of B-NHL (B). The peak concentrations of SF associated with the severity of CRS in the cohort of B-ALL (C) and the cohort of B-NHL (D). (E) The baseline percentage of blasts in BM associated with the severity of CRS in the cohort of B-ALL. (F) The peak concentration of serum C-reactive protein (sCRP) associated with the severity of CRS in the cohort of B-NHL. (G) The international prognostic index (IPI) score at baseline associated with the severity of CRS in the cohort of B-NHL. The peak concentrations of sIL-6 (H) and SF (I), associated with the occurrence of CRES in the cohort of B-ALL. The horizontal lines in each box represent the median values, and the lower and upper borders of each box represent the 25th and the 75th percentiles, respectively, and the whiskers represent the minimum and maximum range.
Furthermore, regardless of causality, fatal AEs within the first month occurred in 3 patients, of whom 1 died of CRS (grade 5) and pneumonia, despite receiving tocilizumab, glucocorticoid, and plasma exchange; 2 died of serious pneumonia owing to infection with cytomegalovirus or Stenotrophomonas maltophilia. Another 9 patients had fatal AEs thereafter, of whom 5 died of infection (supplemental Table 9).
Discussion
Numerous studies have shown that CD19 represents an attractive and effective immunotherapy target for r/r B-ALL/NHL because of its abundant but restricted expression pattern on normal and malignant B cells.34 However, CD19− relapse has emerged as a major challenge for long-term disease control after CD19-directed therapy.12-14 Recently, dual-targeting of CD19 and CD22 has been proposed to overcome CD19− relapse after CAR19 T-cell therapy.23 Similar to CD19, CD22 is expressed in both normal B-cell and associated malignancies,34 including CD19− relapses after CAR19 T-cell therapy.23,24 Its expression begins at the late pre–B-cell stage and is lost during plasma differentiation,34 but is not found on HSCs,34 which makes CD22 one of the most promising targets for B-cell malignancies.23,35 However, diverse constructs, such as dual-signaling CAR or tandem CAR, and cotargeting of multiple tumor-associated antigens expressed on malignant cells are still tested preclinically.22,36,37 Infusion of 2 single-specific CAR T cells may represent a feasible and reliable solution in clinical practice.38 In the present study, we reported the first clinical trial of sequential infusion of CAR19/22 T-cell cocktail to demonstrate its potent clinical efficacy, moderate toxicity and ability to significantly reduce the rate of antigen-escape relapse in patients with r/r B-ALL/NHL. Also, this study first incorporated CAR22 T cell as therapy for B-NHL.
Robust responses were achieved in our study. It achieved an MRD− CR/CRi rate of 96.0% in the B-ALL cohort and a best ORR of 83.3% in the B-NHL cohort. In fact, most of the enrolled patients were characterized by relapsed or refractory diseases, extremely high tumor burden, and unfavorable cytogenetic or genomic aberrations. In particular, antigen-loss relapse was seen in only 1 patient, whereas CD19/CD22–double-positive relapse manifested in almost all the patients with recurrent disease after CAR T-cell infusion, although repeat biopsy and immunophenotyping were conducted in limited cases in the B-NHL cohort, related to sampling difficulty, poor performance status, or patient’s preference. Meanwhile, the severe AEs were mostly cytopenias and the most frequent fatal AE was lung infection, which was attributable in part to the high disease burden and heavy pretreatment of the enrolled patients.39 Nearly all occurrences of high-grade CRS and CRES were reversible and occurred in similar incidences as previously reported.6,8-11,40 Thus, the sequential infusion of the CAR19/22 T-cell cocktail was an efficient and well-tolerated approach to circumventing antigen loss of CD19 or CD22.
The persistence of CAR T cells in vivo is another concern regarding CAR T-cell therapy. CAR19 T cells can persist for months or even years and has the potential to mediate long-term disease control,41 whereas the exhaustion or extinction of CAR19 T cells may result in CD19+ relapse.41-43 Typically, the median disease-free survival achieved in B-ALL patients by CAR19 T-cell therapy is ∼1 year.8-11 In the present study, however, superior long-term survival was not obtained by the infusion of the CAR19/22 T-cell cocktail. The underlying limitation of CAR T-cell persistence may account for this relatively moderate survival benefit, even when used with the third-generation products and cocktail infusion. Indeed, when compared with each single-specific CAR T cell, superior activity or improved survival was not demonstrated by the pooled cocktail of single-specific CAR T cells, but was revealed by dual-specific CAR T cells in a xenograft mice model.22 Therefore, in addition to avoiding antigen-loss relapse, efforts should be made to extend the lifespan of CAR T cells in vivo, if CAR T-cell therapy is to be a definitive therapy rather than a bridging therapy.
The outcome predictive value of specific genetic lesions was investigated in this study, but validation in a multicenter prospective trial is warranted. We found that survival in the cohort of B-ALL did not differ between patients with or without high-risk cytogenetic or genomic aberrations. This observation illustrated a survival benefit in patients with high-risk genetic abnormalities when treated with CAR T-cell therapy and implied that the impact of immunotherapy should be incorporated into the conventional risk stratification of B-ALL. Moreover, considering the comparable PFS between patients with Ph+ ALL or Ph− ALL, the trends toward better OS in Ph+ ALL may be attributable to the administration of tyrosine kinase inhibitors and allo-HSCT in the recurrent patients after CAR T-cell therapy. In addition, similar to previous studies,8,10,11 remission was maintained in most patients with IgH/MYC translocation or in patients with HGBL DH in our study, suggesting that these high-risk patients may benefit from CAR T-cell therapy.
Also, we explored the outcome predictive value of clinical parameters. As reported previously,9 patients with low leukemia burden at baseline had more favorable outcomes and less severity of CRS or incidence of CRES than those with high leukemia burden in the B-ALL cohort. Similarly, survival advantage in the cohort of B-NHL was most pronounced in patients who achieved an overall R3m after CAR T-cell infusion, whereas disease progression tended to be early (within 3 months), which suggested that R3m could represent a useful early predictor of superior survival in B-NHL patients. Moreover, CAR19/22 T-cell cocktail therapy was more effective in patients at first relapse, which could be reflected in future clinical studies to move CAR T-cell therapy forward as a second-line treatment of r/r B-NHL patients.
Taken together, our results indicate that the infusion of CAR19/22 T-cell cocktail is efficient and safe for patients with B-cell malignancies. Dual CD19 and CD22 targeting is a promising approach to reducing antigen-loss relapse in CD19/CD22-directed therapy. The impact of genetic subtypes and clinical parameters on CAR T-cell therapy further underscores the critical importance of optimal patient selection and personalized treatment decisions.
For original data, please contact one of the corresponding authors.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all the faculty and staff of the Clinical and Laboratory Unit (Department of Hematology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology) for clinical and technical support, and Wuhan Bio-Raid Biotechnology Co., Ltd., for cell manufacturing and quality control.
This work is supported by funding from the Key Program of the National Natural Science Foundation of China (grants 81830008 and 81630006) (J.Z.), the National Natural Science Foundation of China (grants 81670152; [L.H.], 81600120 [N.W.], 81570197 [Y.C.], 81873452 [C.L.], and 81873444 [Y.X.]), the Natural Science Foundation of Hubei Province (grants 2018ACA140 and 2016CFA011) (J.Z.), the Huanghe Talents Plan of Wuhan City (grant HHYC-2015002) (J.Z.), the National High Technology Research and Development Program of China (863 Program 2014AA020532) (L.H.), the Milstein Medical Asian American Partnership (MMAAP) Foundation (2018 MMAAP Foundation Hematology Fellowship Award) (L.H.), and the Applied Basic Research Project of Wuhan City (grant 2017060201010156) (Y.X.).
Authorship
Contribution: J.Z. designed and supervised the clinical study; T.Z., C.G., and S.Z. supervised the CAR T-cell production; N.W., C.G., and W.C. conducted preclinical validation and quality control; X.H., W.C., Q.W., J.C., and H.C. collected the clinical data; L.H., N.W., and J.Z. analyzed the data and wrote and revised the manuscript; D.Y., N.W., and L.H. performed the statistical analyses; J.Z., L.H., C.L., Y.X., Y.Z., Y.C., G.W., X.Z., M.Z., N.W., L.J., D.W., and Q.W. enrolled and took care of the patients; and N.W., X.H., L.C., X.M., and Y.L. performed the laboratory tests and monitored the responses of the patients.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jianfeng Zhou, Department of Hematology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Ave, Wuhan, Hubei 430030, China; e-mail: jfzhou@tjh.tjmu.edu.cn; and Liang Huang, Department of Hematology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Ave, Wuhan, Hubei 430030, China; e-mail: lhuang@tjh.tjmu.edu.cn.
