

The genetic and transcriptional program of multiple myeloma has identified novel markers of high-risk disease and mechanisms of pathogenesis. However, there remains a significant gap between prospective identification of high-risk patients and our ability to determine all patients that experience poor outcomes. Epigenetic alterations in myeloma have been less studied, but have significant potential as a translatable biomarker. To better understand how epigenetic programming may contribute to myeloma pathogenesis, we characterized the myeloma DNA methylome.
DNA from CD138+ enriched myeloma specimens from the Multiple Myeloma Research Foundation (MMRF) CoMMpass study (NCT01454297) were obtained after receiving permission from the CoMMpass Tissue Use Committee and Emory IRB. Whole genome bisulfite sequencing (WGBS) was performed by adapter ligation (Kapa HyperPrep) using fully methylated sequencing adapters followed by bisulfite conversion (Qiagen Epitect), library amplification (Kapa Uracil+ HiFi polymerase), and sequencing (NovaSeq 6000 S4 150bp paired-end reads). Sequencing data were mapped with Bismark and CpG methylation calls were extracted using R.
WGBS DNA methylation data was generated for 120 specimens, including 87 baseline, 24 paired relapse, and 9 paired peripheral blood specimens. This yielded ≥5x coverage at 21,393,650 CpGs in 90% of samples with an average coverage of 20x. Myeloma exhibited extensive genomic hypomethylation such that the median level was 42% (range 21-67%) as compared to 64% in plasma cells from healthy individuals. Principle component analysis indicated most variation corresponded with hypomethylation, which occurred in megabase domains devoid of gene expression. In contrast, DNA methylation was retained in the bodies of expressed genes. Principle components 2 and 3 separated samples with t(4;14) translocations from others. This may be due to overexpression of WHSC1 driving excessive histone 3 lysine 36 di-methylation (H3K36me2), which in turn impacts the function of PWWP-domain containing DNA methyltransferases (DNMT3A, DNMT3B).
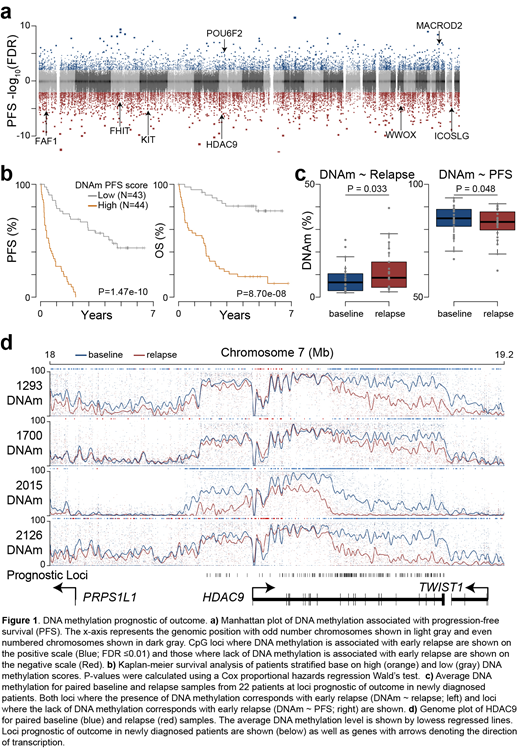
Given the 3-fold variability observed in average methylation, we sought to understand if this was indicative of outcome. Analysis of 87 baseline specimens identified 23,386 loci where DNA methylation (or lack thereof) was prognostic of outcome (FDR ≤0.01) (Figure 1a). These prognostic loci were clustered into contiguous regions often found in gene bodies and could be used to stratify patients by progression-free and overall survival (P-value <1e-7; Hazard ratio >8) (Figure 1b). Importantly, the prognostic value of these CpG were independent of t(4;14) status and ISS stage, indicating these high-risk patients would not have otherwise been identified. Furthermore, analysis of 24 relapse specimens from 22 patients indicated epigenetic remodeling occurred at these prognostic loci. Specifically, loci where the presence of DNA methylation was indicative of poor outcome gained DNA methylation in relapsed samples, and loci where lack of DNA methylation was indicative of poor outcome lost DNA methylation in relapse samples (Figure 1c). These relapse/refractory DNA methylation changes occurred in contiguous regions proximal to genes including PRKCE, MGMT, FHIT, WWOX, and HDAC9 (Figure 1d and data not shown). Cumulatively, these data identify myeloma epigenetic markers of outcome that undergo reprogramming in relapsed samples suggesting they may be indicative of therapeutic resistance.
Lonial:Genentech: Consultancy; Takeda: Consultancy, Research Funding; Amgen: Consultancy; BMS: Consultancy; Janssen: Consultancy, Research Funding; GSK: Consultancy; Karyopharm: Consultancy; Celgene Corporation: Consultancy, Research Funding. Boise:AstraZeneca: Honoraria, Research Funding; Genentech Inc.: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal