

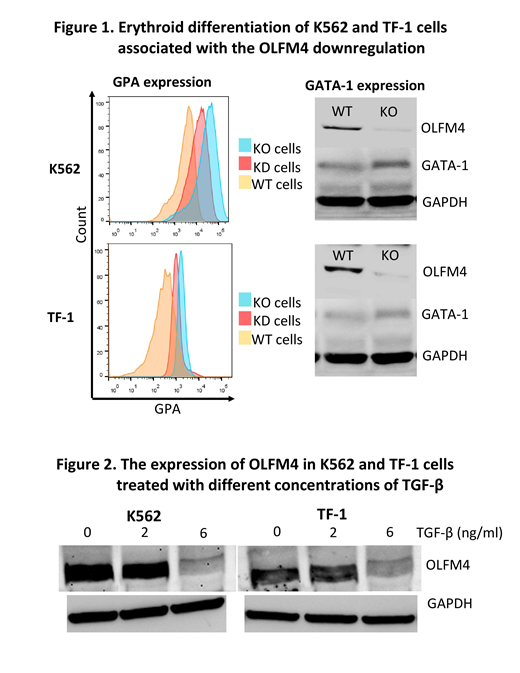
[Background] Olfactomedin 4 (OLFM4), a member of the olfactomedin-related protein family, is constitutively expressed in bone marrow (BM) cells and many gastrointestinal organs and is involved in a variety of cellular functions, including proliferation, differentiation, apoptosis, and cell adhesion in tumor cells. Previous studies have suggested that OLFM4 supports the survival of leukemic stem cells derived from iPS cells of patients with chronic myeloid leukemia. Our recent study revealed a somatic mutation of OLFM4 in HLA allele-lacking granulocytes of a patients with acquired aplastic anemia (AA) in long-term remission (Blood advances, 2(9):1000-1012, 2018). The OLFM4 gene is located at chromosome 13q14.3, which is a commonly deleted region in AA patients with del(13q) who show a good response to immunosuppressive therapy and a high prevalence of increased glycosylphosphatidylinositol-anchored protein(GPI-AP)-deficient cells (Haematologica, 97(12):1845-9, 2012). There may be common mechanisms underlying the preferential commitment between GPI-AP- and del(13q) hematopoietic stem and progenitor cells (HSPCs), like insensitivity to inhibitory cytokines such as TGF-β, in immune-mediated BM failure. To confirm this hypothesis, we studied the effect of OLFM4 knockout on the erythroid differentiation of erythroid leukemia cell lines induced by TGF-β. [Methods] We established OLFM4 knockout (KO) K562 and TF-1 cells using a CRISPR-Cas9 system. OLFM4-knockdown (KD) cells were also prepared using siRNA to validate the results of OLFM4-KO cells. The OLFM4 mRNA and protein levels were determined using quantitative polymerase chain reaction, flow cytometry (FCM), Western blotting, immunocytochemistry, and immunofluorescence methods. The erythroid differentiation was assessed by measuring the expression of glycophorin A (GPA) with FCM, GATA-1 protein expression using Western blotting, and iron staining of the cells. [Results] The OLFM4 KO cells showed slower proliferation than wild-type (WT) cells. Both OLFM4-KO cells and OLFM4-KD cells showed a higher GPA expression than WT cells (median fluorescence intensity [MFI] of K562: 2924 and 2143 vs. 1469 and TF-1: 950 and 870 vs. 694, respectively). OLFM4-KO cells showed erythroid morphology, an elevated expression of GATA-1, and positivity for iron granules, suggesting that OLFM4 KO promoted the erythroid differentiation of K562 and TF-1 cells in RPMI1640 containing 10% fetal bovine serum (Figure 1). When WT cells were cultured in a serum-free culture medium (Steampro34) with or without TGF-β (6 ng/ml) for 8 days, the GPA expression was induced in both TF-1 (MFI: 4358 vs. 883), and K562 cell lines (33440 vs. 25655). The OLFM4 protein levels in these cell lines were significantly decreased by the TGF-β treatment in a dose-dependent manner, suggesting that TGF-β directly downregulated the OLFM4 expression in WT cells; the relative expression of OLFM4 was 1, 0.64, and 0.12 while that of TF-1 was 1, 0.12, and 0.05 at 0, 2, and 6 ng/ml of TGF-β, respectively (Figure 2). [Conclusion] OLFM4 prevents K562 and TF-1 cells from differentiating into erythroid cells in response to TGF-β. The erythroid differentiation of these leukemic cells may be mediated by the downregulation of OLFM4 induced by TGF-β. Haploinsufficiency of OLFM4 due to either a loss of function mutation or del(13q) may be related to the mechanisms underlying the preferential commitment of the mutant HSPCs to erythroid cells in patients with immune-mediated BM failure where TGF-β is abundantly present.
Yoroidaka:Ono Pharmaceutical: Honoraria. Nakao:Takeda Pharmaceutical Company Limited: Honoraria; Novartis Pharma K.K: Honoraria; Kyowa Kirin: Honoraria; Bristol-Myers Squibb: Honoraria; Janssen Pharmaceutical K.K.: Honoraria; Daiichi-Sankyo Company, Limited: Honoraria; Ohtsuka Pharmaceutical: Honoraria; Alaxion Pharmaceuticals: Honoraria; Ono Pharmaceutical: Honoraria; Celgene: Honoraria; Chugai Pharmaceutical Co.,Ltd: Honoraria; SynBio Pharmaceuticals: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal