

Introduction
Mitochondria controls crucial biological pathways such as proliferation, apoptosis and cell growth. However, the implication of mitochondrial activity in the pathogenesis of Multiple Myeloma (MM) still remains unknown and only a few studies connect the mitochondrial status and MM.
We planned to decipher the role of the mitochondria in the MM mechanism of resistance and the potential exploitation of mitochondrial activity as a functional target in the MM therapy.
Methods
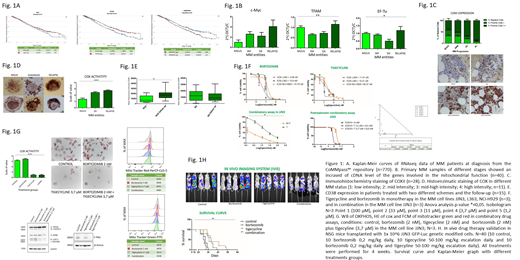
In order to understand the role of mitochondria in MM and its therapeutic exploitation, firstly we explored factors involved in the mitochondrial function (c-Myc, HNRNPK, TFAM, NRF1 and EF-Tu) from 770 MM patients RNAseq CoMMpass℠ data. Furthermore, we performed different studies in our MM 77 patients set: gene expression validation by RT-PCR (n=40), protein expression (COXII) by IHC (n=28); and mitochondrial activity (COX activity) by histoenzymatic-HE assay (n=11). Additionally, we analyzed the impact of bortezomib in the mitochondria regulator CD38 in 50 samples (n=30 RVD, n=20 RD regimens), at diagnosis and 6/9 months follow-up MM patients.
We have tested the effect of tigecycline, a mitochondrial inhibitor, in three regimens: monotherapy, pre-treament of tigecycline (48h) with consecutive bortezomib treatment, and in combination with bortezomib in the MM cell lines JJN3, L363 and NCI-H929. To characterize the molecular mechanisms underlying the cytotoxic effect of tigecycline we analysed mitochondria load and activity (MitoTracker green and red) OXPHOS expression by WB and COX2 activity by HE assay. Finally, we followed an in vivo experiment in NSG mice (n=40) engrafted with the JJN3-GFP cell line (1x106) via tail vein and treated by 4 weeks. Analysis of the in vivo imaging and survival curve were obtained.
Results
The higher expression of factors involved in the mitochondrial function such as: c-Myc, HNRNPK, NRF1 and EF-Tu predict MM poor outcomes (Fig.1A). Furthermore, mitochondrial representative gene and protein expression and activity were found increased in MM relapse stage patients. We showed overexpression of C-Myc, TFAM and EF-Tu on the MM relapsed group (Fig. 1B). Moreover, IHC reveals overexpression of mitochondrial COXII protein in relapse MM patients (p-value ** < 0.001) (Fig. 1C). By functional assays we have demonstrated that gene/protein overexpression drives to an increase of activity (COX HE) in MM at relapse (p-value ***< 0.0001). (Fig. 1D). Moreover, we observed an increase of CD38 expression in patients with RVD regimen, but not without bortezomib (RD regimen) (Fig. 1E). Together these results suggest elevation of mitochondrial activity plays a role in the mechanism of resistance to treatment and/or progression of MM and the consequent relapse of the patients.
In vitro studies with tigecyline and bortezomib showed cytotoxic effects in three MM cell lines (IC50 JJN3 11,91 µM; IC50 L363 10,21 µM and NCI-H929 26,37 µM, p-value *< 0.05). Moreover, bortezomib and tigecyline showed high levels of synergism (CI 0,19) (Fig. 1F). In fact, the "conditioning" treatment with tigecyline revert the resistance to bortezomib. The cells treated with tigecycline reflect diminishing in the mitochondria respiration by MitoTracker assays, decrease of COX activity and respiratory chain complexes, suggesting a reduction of mitochondrial activity (Fig. 1G). These molecular effects are exacerbated by the tigecycline and bortezomib combination. However, bortezomib monotherapy not decrease or inclusive, increase, all the molecular mechanisms of mitochondria studied. Finally, mice groups treated with tigecycline alone or in combination with bortezomib reported a better survival and lower JJN3-GFP infiltration (p-value *< 0.05) (Fig. 1H).
Conclusion
To sum up, these findings highlight new vulnerabilities in myeloma cells, suggesting a potential therapeutic target in the treatment of the disease. The metabolic activation of myeloma cells with the collaboration of CD38 and/or c-Myc overexpression or his regulators (e.g. HNRNPK) in response to bortezomib treatment lead an increase of mitochondria respiration. These data confirm the important role of mitochondria in the loss of efficacy in inhibitors of proteasome treatment. Thus, mitochondrial respiration emerges as a novel target in bortezomib relapsed MM patients, and, potentially, in multiple c-Myc, HNRNPK and CD38 overexpression neoplasms.
Munshi:Adaptive: Consultancy; Oncopep: Consultancy; Janssen: Consultancy; Takeda: Consultancy; Amgen: Consultancy; Celgene: Consultancy; Abbvie: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal