

B cell receptor (BCR) signals generated by chronic interactions with external autoantigens or cell-autonomous BCR-BCR interactions play a critical role in the pathogenesis of CLL (Dühren-von Minden M et al, Nature 2012; Iacovelli S et al, Blood 2015). However, the role of these signals in regulating CLL cell responses and in particular in regulating CLL cell proliferation has still not been established. To address this issue, we first reanalyzed a previously generated dataset of human CLL cells stimulated with immobilized anti-IgM (Dal Bo M et al, Oncotarget 2015), which revealed differential expression of a number of genes involved in regulating the G1 phase of the cell cycle. The observed changes included upregulation of positive regulators of the cell cycle, such as CCND1, CCND2 and CDK4, but also upregulation of negative regulators, such as the cell cycle inhibitors CDKN1A, CDKN2A and CDKN2B. Changes in the expression of these genes were further validated by RQ-PCR analysis of a separate set of anti-IgM-stimulated human CLL cells (n=12), as well as by analysis of murine Eμ-TCL1-derived CLL cells stimulated with their cognate antigen phosphatidylcholine (PtC) (n=5). To investigate whether these genes are also induced by cell-autonomous BCR-BCR interactions, we analyzed the same samples following treatment with the SYK inhibitor entospletinib or the BTK inhibitor ibrutinib (1 μM each for 20 hours). A modest reduction in the levels of the cell cycle inhibitor CDKN2B but no change in the levels of any of the positive cell cycle regulators was observed, suggesting that only signals generated by external antigens can induce CLL cells to enter the cell cycle.
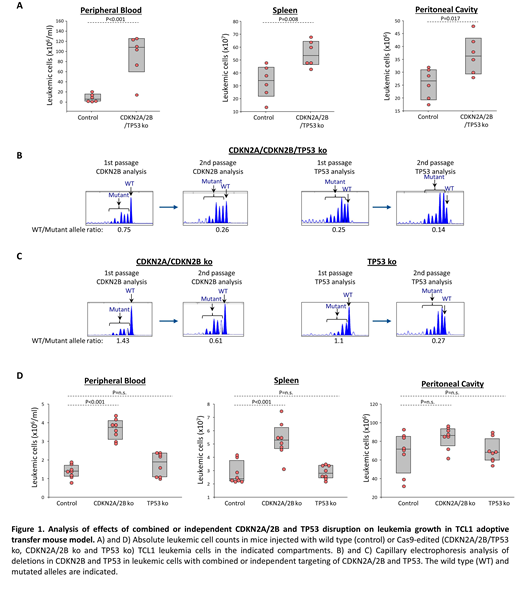
The finding that the negative regulators CDKN1A, CDKN2A and CDKN2B are induced upon BCR engagement with external ligand appeared particularly interesting, considering that these genes are frequently downregulated in cases with Richter transformation because of co-existing genetic lesions in CDKN2A/2B and TP53, which is a key transcriptional activator of CDKN1A. Moreover, these findings suggested that genetic lesions in CDKN2A, CDKN2B and TP53 could contribute to Richter transformation by increasing the proliferation of leukemic cells stimulated with external autoantigen. To further address this possibility, we simultaneously targeted CDKN2A, CDKN2B and TP53 by CRISPR/Cas9 in primary Eμ-TCL1-derived murine CLL cells expressing an autoreactive BCR with specificity for the PtC autoantigen. The Cas9-edited and non-edited CLL cells were then expanded in immunodeficient NSG mice and were subsequently transferred in syngeneic recipients (n=6-8 per group) to evaluate the effects of CDKN2A/2B/TP53 knockdown on leukemia growth and proliferation. Combined targeting of CDKN2A, CDKN2B and TP53 resulted in accelerated leukemia growth, as evidenced by the significantly higher number of leukemic cells in the peritoneal cavity, peripheral blood and spleen of mice that received Cas9-edited vs wild type CLL cells (Figure 1A). The Cas9-edited leukemic cells showed markedly reduced expression of CDKN1A and CDKN2B protein (CDKN2A expression was not assessed in these experiments) and significantly higher expression of the proliferative markers Ki67, BrdU and phospho-histone H3 compared to non-edited leukemic cells. Moreover, indel analysis by amplicon capillary electrophoresis showed an increase in the ratio of CDKN2B mutant and TP53 mutant alleles vs wild type alleles during in vivo propagation, further suggesting that cells with combined CDKN2A/2B/TP53 deficiency have a growth advantage over wild type CLL cells (Figure 1B). A separate experiment with independent knockdown of CDKN2A/2B and TP53 also revealed selection of the mutant alleles during in vivo propagation, although only CDKN2A/2B knockdown resulted in a significant increase in tumor burden (Figure 1C and 1D). In conclusion, these data demonstrate that BCR interactions with external autoantigens induce both positive and negative regulators of the cell cycle and suggest that genetic lesions associated with Richter transformation cooperate with BCR signals by downregulating the negative regulators and facilitating cell cycle progression.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal