

Introduction:RAS-activating mutations are common in both childhood B- and T-acute lymphoblastic leukemia (ALL). Prior studies of B- and T-ALL have shown that RAS mutations become enriched during treatment in minimum residual disease-positive cases, are associated with a poor glucocorticoid response, and are associated with inferior survival in relapsed disease. There is a need for more tractable preclinical models of RAS mutation-driven B- and T-ALL. Currently, most genetically-engineered mouse models generated to study KRASG12D-driven ALL have a high latency, low penetrance, and/or necessitate the use of multiple technical manipulations, which can yield inconsistent results. Here, we set out to generate a more efficient and penetrant mouse model of KRASG12D-driven B-ALL or T-ALL.
Methods:We utilized KRASLSL-G12D/+ mice, which carry KRASG12D preceded by a Lox-Stop-Lox (LSL) site. KRASLSL-G12D/+ males were crossed with Mb1Cre/+ females, which express Cre in most (68-90%) B lineage cells and in a small percentage (<1%) of T cells. This yielded controls and genotypes with both the KRASLSL-G12DandCre genes to drive KRASG12D expression in B and T cells.
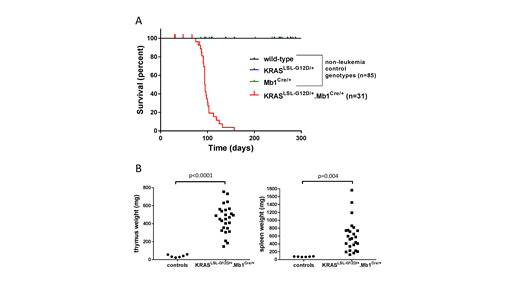
Results:KRASLSL-G12D/+Mb1Cre/+ mice developed T-ALL with a median latency of 101 days (range 75-157 days old) and 83.8% penetrance (Figure 1A). A few mice were censored due to early deaths, before they would have likely developed T-ALL, from non-leukemic complications typically involving malocclusion. No mice developed B-ALL. Flow cytometry demonstrated expansion of CD4+CD8+ cells in peripheral blood, bone marrow, and/or spleen in 8 of the 9 mice assayed. One mouse had CD8+ T-ALL. Leukemic mice had significantly increased thymus weight (453 vs 40 mg, p < 0.0001) and spleen weight (601 vs 76 mg, p = 0.004) compared to age-matched (86-139 days old) control mice (Figure 1B).
Conclusions: Here we describe a short latency, high penetrance mouse model of KRASG12D-driven T-ALL. No mice developed B-ALL, perhaps because the small percentage of T cells expressing Cre gained a greater proliferative advantage relative to any effects manifested in B cells. KRAS mutations are present in 5-10% of pediatric T-ALL cases at diagnosis and 12% at relapse, and predict poor response to therapy and poor survival in relapsed cases. Thus, this constitutes a useful model for studying a clinically relevant disease. Currently, almost all genetically-engineered models of RAS mutation-driven T-ALL involve technical manipulation, including injections with pIpC or retrovirus, and/or secondary transplantation into irradiated mice; or exhibit a much longer latency to disease (eg a KRASLSL-G12D model with Cre driven by the Lck promoter, with median disease latencies of 121 and 180 days in two prior reports). This model may be utilized to study candidate therapies for KRAS mutation-driven T-ALL. Because this model does not involve irradiation, it is also better suited for studies of T-ALL in the native host microenvironment, and for studies of pre-leukemic evolution, including interactions with the host immune system.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal