

Constitutional GATA2 deficiency caused by heterozygous germline GATA2 mutations has a broad spectrum of clinical phenotypes including systemic infections, lymphedema, cytopenias, myelodysplasia, and a high risk of developing myeloid leukemias. GATA2 deficiency is recognized as a major MDS predisposition syndrome, germline GATA2 mutations are found in 7% of primary MDS cases in children and adolescents. Monosomy 7 and trisomy 8 are frequent chromosomal abnormalities in GATA2 deficiency and features of disease prognosis and malignant transformation. GATA2 mutations mostly affect two zinc finger domains and broadly classify into three major categories: missense, null, and regulatory region mutations. Profound monocytopenia, B-cell and NK-cell lymphopenias, and dendritic cell deficiency are specific for GATA2 deficiency. Absence of multi-lymphoid progenitor and decreased granulocyte -macrophage progenitors occur early in disease. There are limited studies of gene expression profiles in GATA2 deficiency and these usually in bulk populations, partly due to the paucity of cells in patients. How GATA2 deficiency negatively affects hematopoiesis, especially the preferential loss of specific lineages, is poorly understood.
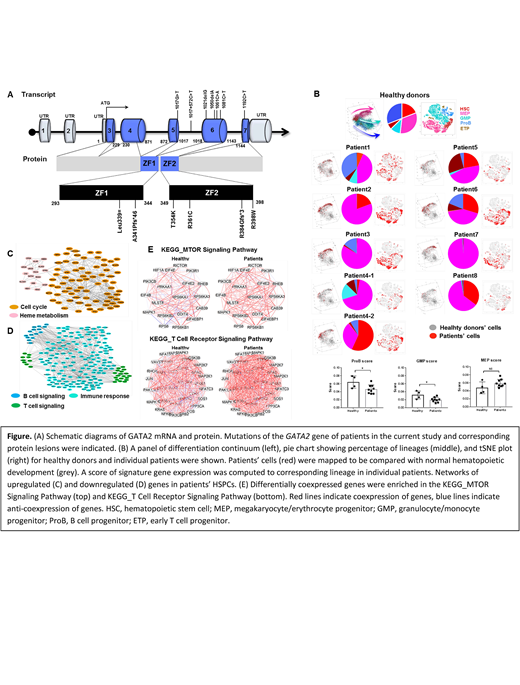
We performed single-cell RNA sequencing of sorted bone marrow CD34+ hematopoietic stem and progenitor cells (HSPCs) from eight GATA2 deficiency patients, who had various well characterized pathogenic GATA2 mutations and clinically manifest myelodysplasia (Fig A). We characterized transcriptomes in lineages, computationally defined cells with chromosomal abnormalities, and described gene expression of these cells. We first used gene expression signatures of single HSPCs from healthy donors to impute identity of stem cells or progenitors of erythroid/megakaryocytic (MEP), myeloid and lymphoid lineages, and then proceeded to reconstruct hematopoietic differentiation by pseudotemporal ordering. Mapping patients' cells onto normal hematopoiesis, we observed marked deficiency in granulocyte/monocyte/lymphocyte differentiation and relatively preserved MEP (Fig B, top). Based on signature genes of each differentiated lineage, we computed a score of gene expression to corresponding lineage in individual patient and found a significantly lower score for granulocyte/monocyte and B cell progenitors in patients, MEP scores tended to be higher in patients without significance (Fig B, bottom). Upregulated genes in patients' HSPCs related to erythroid differentiation, and cell cycle and proliferation (Fig C), while downregulated gene sets were highly enriched in immune responses (Fig D). Differential co-expressed genes were defined by weighted correlation network analysis; they were enriched in immune response and DNA repair (genes exhibited higher correlation in MTOR and T cell receptor signaling pathway in patients [Fig E]). We observed differential effects of GATA2 mutations in distinct lineages, which could only be resolved with the single cell method in rare cell populations. In HSCs, upregulated genes were related to apoptosis and downregulated genes were involved in maintenance of quiescence and cell cycle; there was increased expression of erythroid/megakaryocytic priming programs and decreased expression of lymphoid priming programs. We also observed lower GATA2 expression levels at initial stages of hematopoiesis, likely contributing to disturbed stem cell homeostasis in GATA2 deficiency. DNA repair genes were downregulated in trisomy 8 cells, possibly rendering these cells vulnerable to second-hit somatic mutations and additional chromosomal abnormalities. Cells with complex cytogenetics had defects in multi-lineage differentiation and cell cycle.
In conclusion, germline GATA2 mutations modulate gene expression and change gene coexpression patterns. Distinct lineages show different transcriptional profiles resulting from GATA2 mutations. The prominent deficiency in myeloid/lymphoid lineages in GATA2 deficiency is partly due to expression of aberrant gene programs in HSCs prior to lineage commitment.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal