

Introduction
FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutations are found in approximately one quarter of acute myeloid leukemia (AML) cases. Its presence results in constitutive activation of the FLT3 receptor tyrosine kinase and its downstream growth/pro-survival pathways including MAPK/ERK, PI3K/AKT, and JAK/STAT, and confers a poor prognosis. Gilteritinib is a selective inhibitor of FLT3 recently approved by the Food and Drug Administration for treatment of patients with relapsed/refractory AML and a FLT3 mutation. However, gilteritinib exposure induces upregulation of FLT3 - a mechanism of resistance. Previously, we showed that CUDC-907, a dual PI3K/histone deacetylase inhibitor, downregulates FLT3 expression (Li X, et al. Haematologica. 2019; epub ahead of print). We therefore hypothesized that combining CUDC-907 with gilteritinib would abrogate FLT3 upregulation and expression, resulting in synergistic antileukemic activities against FLT3-mutated AML.
Methods
FLT3-ITD AML cell lines and primary patient samples were treated with gilteritinib or CUDC-907, alone or in combination at clinically achievable concentrations, and subjected to annexin V/propidium iodide staining and flow cytometry analysis to quantify apoptosis. Protein levels of FLT3, Bcl-2 family proteins, and key components of the MAPK/ERK, PI3K/AKT, and JAK/STAT pathways were examined using western blotting. The impact of the observed alterations upon apoptosis were confirmed via overexpression, knockdown, and targeted inhibitor experiments. Real-time RT-PCR was used to determine FLT3 transcript levels. The FLT3-ITD AML cell line MV4-11 was used to generate a xenograft mouse model to assess in vivo efficacy of the two agents.
Results
CUDC-907 and gilteritinib demonstrated potent synergistic antileukemic effects in FLT3-ITD AML cell lines in vitro and patient samples ex vivo, with combined therapy. CUDC-907 abolished gilteritinib-induced expression of FLT3 in both cell lines and primary patient samples. Gilteritinib treatment reduced p-AKT, p-S6, and p-STAT5 and increased p-ERK, while CUDC-907 reduced p-AKT and p-ERK, and upregulated p-STAT5. The combination of gilteritinib and CUDC-907 decreased not only p-AKT and p-S6, but also p-ERK and p-STAT5. Targeted inhibition of ERK and JAK2/STAT5 signaling by SCH772984 and AZD1480, respectively, confirmed their roles in resistance to gilteritinib and CUDC-907 monotherapies, respectively. Combined gilteritinib and CUDC-907 treatment reduced expression of the anti-apoptotic BCL-2 family member Mcl-1 and increased expression of the pro-apoptotic protein Bim. MCL-1 overexpression and BIM knockdown partially rescued FLT3-ITD AML cells upon drug treatment, confirming their role in the antileukemic activity of combined gilteritinib and CUDC-907.
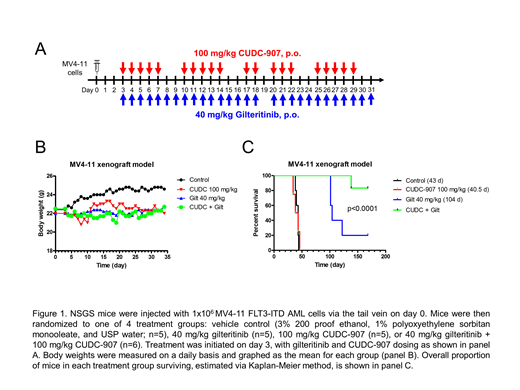
To determine in vivo efficacy of the two agents, NSGS mice were injected with MV4-11 cells. Three days later, the mice were randomized into vehicle control (n=5), 40 mg/kg gilteritinib (oral gavage; n=5), 100 mg/kg CUDC-907 (oral gavage; n=5) or combination (40 mg/kg gilteritinib + 100 mg/kg CUDC-907; n=6) groups. CUDC-907 was given daily for 5 days on, 2 days off, for a total of 4 cycles. Gilteritinib was administered daily for 28 days. Both agents were well tolerated; maximal weight loss was 5.5%, 0.9%, and 6.7% in the CUDC-907, gilteritinib, and combination groups, respectively. Median survival of mice in the vehicle control group was 43 days. Median survival in the CUDC-907 monotherapy and gilteritinib monotherapy arm was 40.5 days and 104 days, respectively. One mouse in the combination therapy arm died on day 138, while the remaining 5 mice in the combination therapy arm continue to survive, as of time of writing (day 168), and are asymptomatic (Figure 1).
Conclusion
We confirmed that the combination of CUDC-907 plus gilteritinib synergistically induces apoptosis in both FLT3-ITD AML cell lines and primary patient samples, and that gilteritinib-induced FLT3 expression is abolished by CUDC-907. Cooperative inhibition of the PI3K-AKT, JAK-STAT, and RAS-RAF pathways, as well as upregulation of Bim/downregulation of Mcl-1 all appear to contribute to this observed antileukemic synergy. Our cell line-derived xenograft mouse model provides strong evidence of in vivo efficacy and robust grounds for clinical translation of this therapeutic combination.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal