

Key Points
Subcutaneously administered daratumumab had similar safety and PK profile, and lower infusion-related reactions, compared with the IV formulation.
The 1800-mg subcutaneous dose of daratumumab induced deep, durable responses in patients with heavily pretreated MM.

Abstract
Daratumumab, a human monoclonal antibody targeting CD38, is approved as monotherapy and in combination regimens for patients with multiple myeloma (MM). Currently, daratumumab is administered IV. The phase 1b PAVO (MMY1004) study evaluated subcutaneously administered daratumumab in combination with the recombinant human hyaluronidase PH20 enzyme (rHuPH20) in patients with relapsed or refractory MM. Part 1 of the study, reported here, evaluated a mix-and-deliver (MD) formulation of daratumumab and rHuPH20 (DARA-MD) administered by subcutaneous infusion. Patients received subcutaneous daratumumab according to the approved IV monotherapy dosing schedule at 1200 mg (n = 8) or 1800 mg (n = 45). Primary end points were safety and pharmacokinetic (PK) variables. The most common treatment-emergent adverse events with DARA-MD 1200 mg were thrombocytopenia, upper respiratory tract infection, insomnia, and decreased appetite (37.5% each). Anemia (33.3%), upper respiratory tract infection, pyrexia, and diarrhea (26.7% each) were the most common treatment-emergent adverse events with DARA-MD 1800 mg. One patient in the 1200-mg dose group (12.5%) and 11 patients in the 1800-mg dose group (24.4%) experienced infusion-related reactions, which were generally grade 1/2 and typically occurred at the first infusion. The 1800 mg dose achieved similar or greater serum concentrations compared with the 16 mg/kg IV dose. Overall response rates of 25.0% and 42.2% were achieved with 1200-mg and 1800-mg DARA-MD, respectively. Subcutaneous administration of DARA-MD was well tolerated in patients with relapsed or refractory MM, with the 1800-mg dose exhibiting PK concentrations and responses consistent with IV daratumumab in a similar patient population. This study was registered at www.clinicaltrials.gov as #NCT02519452.
Introduction
Daratumumab is a human immunoglobulin G1 kappa monoclonal antibody targeting CD38 with direct on-tumor and immunomodulatory mechanisms of action.1 Daratumumab-based combinations have consistently shown unprecedented efficacy in multiple myeloma (MM) across all lines of therapy, leading to the approval of daratumumab as monotherapy and in combination with standard-of-care regimens for the treatment of MM.2-7
In clinical studies, the median duration of the first, second, and subsequent IV daratumumab infusions of 1000 mL (first) or 500 mL (subsequent) were 7.0, 4.3, and 3.4 hours, respectively.8 Although daratumumab has consistently shown tolerability across clinical studies, infusion-related reactions (IRRs) are observed in ∼50% of patients.2-8 IRRs observed with daratumumab are generally mild to moderate, manageable, and occur primarily earlier in treatment, with 40% of patients experiencing an IRR at the first infusion, 2% at the second infusion, and 4% at all subsequent infusions.8 The gradual absorption of daratumumab into systemic circulation after subcutaneous administration may reduce the incidence of IRRs and improve tolerability. Subcutaneous administration of daratumumab may also improve convenience for both patients and health care providers by reducing infusion time.
Agents injected subcutaneously must traverse the interstitial matrix of the skin before systemic absorption occurs. The structure and composition of this matrix limit the injection volume at each site to ∼1 to 2 mL.9 Recombinant human hyaluronidase PH20 (rHuPH20) depolymerizes hyaluronan in the subcutaneous space, leading to an increase in bulk fluid flow that facilitates the dispersion and absorption of injected drugs at more rapid infusion rates.9,10 This local and transient action within the subcutaneous space facilitates drug administration. The feasibility of administering anticancer agents subcutaneously in combination with rHuPH20 has been reported for trastuzumab and rituximab, leading to their approval in Europe and the United States.11-14
Based on the infusion time and incidence of IRRs associated with daratumumab, a subcutaneous delivery method for daratumumab that significantly shortens the duration of infusion without compromising the safety or efficacy of the drug is desirable. The current report describes part 1 of the first study (MMY1004; PAVO) to assess the safety, pharmacokinetic (PK) variables, and antitumor activity of subcutaneous delivery of the mix-and-deliver (MD) formulation of daratumumab in combination with rHuPH20 (hereafter referred to as DARA-MD) in patients with relapsed or refractory MM (RRMM).
Methods
Study design and patients
PAVO (MMY1004) was a phase 1b, open-label, multicenter, dose-escalation, 2-part study evaluating the safety and PK profile of subcutaneous daratumumab. Part 1, reported here, was a dose escalation study evaluating DARA-MD administered subcutaneously at doses of 1200 or 1800 mg. Part 2 is ongoing and will evaluate a concentrated coformulation of daratumumab and rHuPH20 at the dose identified in part 1. Eligible patients were aged ≥18 years, with a documented diagnosis of MM (according to International Myeloma Working Group criteria15 ), measurable serum or urine M-protein levels, and an Eastern Cooperative Oncology Group (ECOG) performance status ≤2. Patients with RRMM received ≥2 previous lines of treatment, including a proteasome inhibitor and an immunomodulatory drug, and were naive to anti-CD38 therapy. Eligible patients had hemoglobin levels ≥7.5 g/dL, absolute neutrophil count ≥1.0 × 109/L, platelet count ≥75 × 109/L (patients in whom <50% of bone marrow nucleated cells were plasma cells; otherwise, ≥50 × 109/L), aspartate aminotransferase level ≤3.0 × upper limit of normal (ULN), alanine aminotransferase level ≤3.0 × ULN, creatinine clearance ≥20 mL/min/1.73 m2, corrected serum calcium ≤14 mg/dL, and total bilirubin ≤2.0 × ULN (≤1.5 × ULN in patients with congenital bilirubinemia).
In part 1, daratumumab was mixed with rHuPH20 (ENHANZE drug delivery technology; Halozyme, Inc, San Diego, CA) and administered subcutaneously at 2 dose levels by using a sequential enrollment strategy. Population PK analyses and simulations for daratumumab determined that both weight-based and flat dosing protocols are reasonable approaches for administering daratumumab. To simplify dose administration and reduce drug wastage compared with weight-based dosing, a flat-dose approach was chosen for daratumumab at dose levels of 1200 mg and 1800 mg. The 1200-mg dose of DARA-MD is equivalent to the amount of drug administered using the approved DARA 16 mg/kg IV dose in a 75-kg patient. The 1800-mg dose of DARA-MD, selected in the event that the bioavailability of the 1200-mg dose was <100%, is equivalent to the amount of drug administered with DARA 24 mg/kg IV in a 75-kg patient, which was well tolerated in GEN501 part 1.2 In PAVO, group 1 received a mixture of DARA 1200 mg and rHuPH20 30 000 U in a total volume of 60 mL over ∼20 minutes. Group 2 received DARA 1800 mg and rHuPH20 45 000 U, administered in a total volume of 90 mL over ∼30 minutes. DARA-MD was administered by subcutaneous infusion using a syringe pump and was delivered in the right or left abdominal wall, alternating sites between doses. Treatment was administered in 28-day cycles, once weekly in cycles 1 and 2, every 2 weeks in cycles 3 to 6, and every 4 weeks thereafter until disease progression or unacceptable toxicity.
Patients received preinfusion and postinfusion medications to reduce the incidence of IRRs, which are described in detail in the supplemental Appendix (available on the Blood Web site). Safety was reviewed by a study evaluation team (composed of all investigators, the medical monitor, clinical pharmacologist, and statistician from the sponsor) after completion of cycle 1 to evaluate dose-limiting toxicities and to determine the dose for cohort 2. The study evaluation team also reviewed PK data after completion of cycle 3 day 1 to support dose selection decisions during part 1. In the absence of dose-limiting toxicities during cycle 1 for >2 patients in group 1 (of ≥6 toxicity-evaluable patients), dose escalation was permitted to the next dose level (group 2, DARA-MD 1800 mg) with the same dosing schedule as group 1.
Blood samples were collected for PK profiles of the first dose (cycle 1 day 1 before dosing, end of infusion [EOI], EOI + 2 hours, and EOI + 12 hours); days 2, 3, 4, and 8; and the last weekly dose (cycle 2 day 22 predose, EOI, EOI + 2 hours, and days 23 and 25, and cycle 3 day 1 predose). To define trough concentrations, PK samples were collected before drug administration on cycle 1 days 8, 15, and 22; cycle 2 days 1, 8, 15, and 22; and day 1 of cycles 3, 4, 6, and 8. Samples were analyzed for anti-daratumumab and anti-rHuPH20 antibodies (immunogenicity) before dosing on days 1 and 15 of cycle 1; day 22 of cycle 2; day 1 of cycle 4; and posttreatment weeks 4 and 8. Samples for PK analysis and immunogenicity were also obtained at 4 and 8 weeks after the final dose of study medication.
End points, assessments, and statistical analyses
Primary end points were the PK variables and safety of DARA-MD. Secondary end points included immunogenicity of daratumumab and rHuPH20, overall response rate (ORR; defined as partial response [PR] or better), rate of complete response (CR), time to response, and duration of response.
The primary PK end point, trough concentration (Ctrough) at the end of weekly dosing (before the cycle 3 day 1 dose16 ), and all other PK analyses were evaluated in patients who received ≥1 dose of study drug and provided ≥1 postinfusion PK sample. The incidence of anti-daratumumab antibodies and the incidence and baseline prevalence of anti-rHuPH20 antibodies were assessed for all patients who received ≥1 dose of DARA-MD and had appropriate samples.
The safety population included all patients who received ≥1 dose of the study drug. Safety assessments included adverse events (AEs), physical examinations, electrocardiograms, photographs of subcutaneous infusion sites, infusion site evaluations, clinical laboratory parameters, vital sign measurements, and ECOG performance status. AEs were assessed by using National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03.17 Dose-limiting toxicities included grade 3 (unresolved with management) or 4 IRRs, grade ≥3 nonhematologic AEs (except for grade 3 nausea or diarrhea that responded to treatment, grade 3 fatigue, isolated grade 3 γ-glutamyltransferase elevation, tumor lysis syndrome, or hyperuricemia), and grade 4 hematologic AEs (thrombocytopenia, neutropenia, febrile neutropenia, or anemia). All patients had their blood types assessed before their first daratumumab administration and were encouraged to carry a card indicating their blood type for the duration of the study and for 6 months after the last dose received.
Responses were evaluated at the beginning of each treatment cycle and were assessed according to International Myeloma Working Group criteria.15 Duration of response was calculated from the time of initial response (PR or better) until disease progression and time to response was calculated from the first dose of DARA-MD to achievement of PR or better.
Study oversight
The study was registered with clinicaltrials.gov (#NCT02519452). The research was approved by the clinical study sites’ institutional review boards or ethics committees, and all patients gave written informed consent. The study design and analyses were devised by the investigators and sponsor.
Statistical analyses
No formal statistical hypothesis testing was conducted; all outcomes were summarized by using descriptive statistics only. The sample size was determined by the total number of patient cohorts and cohort size (∼8 patients per cohort).
Results
Patients and treatment
The first patient was enrolled into the study on 23 October 2015. At the clinical cutoff date of 27 February 2018, eight patients were enrolled into the 1200-mg dose group and 45 patients were enrolled into the 1800-mg group (interim safety and PK analyses of the first 10 patients were followed by cohort expansion, with an additional 35 patients enrolled at the 1800-mg dose level). Patient demographic and clinical characteristics were largely comparable between dosing groups (Table 1). For patients in the DARA-MD 1200-mg and 1800-mg groups, the median age was 65.5 and 63.0 years, respectively. Only 1 patient in each dose group had a baseline ECOG performance status of 2. Patients who received DARA-MD 1200 mg had a median (range) of 5 (2-10) previous lines of therapy over a median 6.6 years since diagnosis vs 4 (2-11) and 5.9 years for patients who received the 1800-mg dose. The majority of patients in both the 1200-mg and 1800-mg dose groups were refractory to both a proteasome inhibitor and an immunomodulatory drug (62.5% vs 64.4%) or the last previous line of therapy (87.5% vs 80.0%). In the 1200- and the 1800-mg dose groups, 62.5% and 82.2% of patients had received a previous autologous stem cell transplantation.
Baseline demographic and clinical characteristics
| Characteristic | 1200 mg (n = 8) | 1800 mg (n = 45) |
|---|---|---|
| Age, y | ||
| Median (range) | 65.5 (49-78) | 63.0 (36-79) |
| ≥75 y, n (%) | 1 (12.5) | 4 (8.9) |
| Weight, median (range), kg | 75.0 (53.0-82.5) | 74.8 (48.0-133.0) |
| Baseline ECOG status, n (%) | ||
| 0 | 2 (25.0) | 11 (24.4) |
| 1 | 5 (62.5) | 33 (73.3) |
| 2 | 1 (12.5) | 1 (2.2) |
| ISS stage at screening, n (%)* | ||
| N | 6 | 45 |
| I | 1 (16.7) | 21 (46.7) |
| II | 3 (50.0) | 15 (33.3) |
| III | 2 (33.3) | 9 (20.0) |
| Time since diagnosis, median (range), y | 6.55 (1.9-10.3) | 5.94 (1.1-15.2) |
| IgG myeloma, n (%) | 3 (37.5) | 30 (66.7) |
| Previous lines of therapy, n (%) | ||
| Median (range) | 5 (2-10) | 4 (2-11) |
| ≤3 | 3 (37.5) | 16 (35.6) |
| >3 | 5 (62.5) | 29 (64.4) |
| Previous ASCT, n (%) | 5 (62.5) | 37 (82.2) |
| Previous IMiD, n (%) | 8 (100) | 45 (100) |
| Previous lenalidomide, n (%) | 8 (100) | 45 (100) |
| Previous PI, n (%) | 8 (100) | 45 (100) |
| Previous bortezomib | 8 (100) | 44 (97.8) |
| Refractory to, n (%) | ||
| PI only | 0 | 1 (2.2) |
| IMiD only | 1 (12.5) | 7 (15.6) |
| Both PI and IMiD | 5 (62.5) | 29 (64.4) |
| Last line of therapy | 7 (87.5) | 36 (80.0) |
| Cytogenetic risk, n (%)† | ||
| N | 8 | 37 |
| Standard risk | 4 (50.0) | 30 (81.1) |
| High risk | 4 (50.0) | 7 (18.9) |
| del17p | 3 (37.5) | 5 (13.5) |
| t(4;14) | 2 (25.0) | 3 (8.1) |
| t(14;16) | 0 | 0 |
| Characteristic | 1200 mg (n = 8) | 1800 mg (n = 45) |
|---|---|---|
| Age, y | ||
| Median (range) | 65.5 (49-78) | 63.0 (36-79) |
| ≥75 y, n (%) | 1 (12.5) | 4 (8.9) |
| Weight, median (range), kg | 75.0 (53.0-82.5) | 74.8 (48.0-133.0) |
| Baseline ECOG status, n (%) | ||
| 0 | 2 (25.0) | 11 (24.4) |
| 1 | 5 (62.5) | 33 (73.3) |
| 2 | 1 (12.5) | 1 (2.2) |
| ISS stage at screening, n (%)* | ||
| N | 6 | 45 |
| I | 1 (16.7) | 21 (46.7) |
| II | 3 (50.0) | 15 (33.3) |
| III | 2 (33.3) | 9 (20.0) |
| Time since diagnosis, median (range), y | 6.55 (1.9-10.3) | 5.94 (1.1-15.2) |
| IgG myeloma, n (%) | 3 (37.5) | 30 (66.7) |
| Previous lines of therapy, n (%) | ||
| Median (range) | 5 (2-10) | 4 (2-11) |
| ≤3 | 3 (37.5) | 16 (35.6) |
| >3 | 5 (62.5) | 29 (64.4) |
| Previous ASCT, n (%) | 5 (62.5) | 37 (82.2) |
| Previous IMiD, n (%) | 8 (100) | 45 (100) |
| Previous lenalidomide, n (%) | 8 (100) | 45 (100) |
| Previous PI, n (%) | 8 (100) | 45 (100) |
| Previous bortezomib | 8 (100) | 44 (97.8) |
| Refractory to, n (%) | ||
| PI only | 0 | 1 (2.2) |
| IMiD only | 1 (12.5) | 7 (15.6) |
| Both PI and IMiD | 5 (62.5) | 29 (64.4) |
| Last line of therapy | 7 (87.5) | 36 (80.0) |
| Cytogenetic risk, n (%)† | ||
| N | 8 | 37 |
| Standard risk | 4 (50.0) | 30 (81.1) |
| High risk | 4 (50.0) | 7 (18.9) |
| del17p | 3 (37.5) | 5 (13.5) |
| t(4;14) | 2 (25.0) | 3 (8.1) |
| t(14;16) | 0 | 0 |
ASCT, autologous stem cell transplantation; IgG, immunoglobulin G; IMiD, immunomodulatory drug; ISS, International Staging System; PI, proteasome inhibitor.
ISS staging was derived based on the combination of serum β2-microglobulin and albumin.
Cytogenetic abnormalities were based on fluorescence in situ hybridization or karyotype testing.
At the clinical cutoff date, patients in the 1200-mg group had a median (range) duration of 5.2 (1.6-13.9) months of follow-up compared with a median 8.3 (1.8-22.3) months of follow-up among patients in the 1800-mg group. Among patients who received DARA-MD 1200 mg, all 8 (100%) patients discontinued treatment, a result of progressive disease (5 [62.5%]), physician decision (1 [12.5%]), patient withdrawal (1 [12.5%]), or AEs (1 [12.5%]). In the DARA-MD 1800-mg group, 37 (82.2%) patients discontinued treatment, a result of progressive disease (30 [66.7%]), physician decision (5 [11.1%]), patient withdrawal (1 [2.2%]), and death (1 [2.2%]).
PK properties and immunogenicity
The mean daratumumab serum concentration profiles for subcutaneous daratumumab after the first dose and the last weekly dose (eighth planned dose, which was to be administered on cycle 2 day 22) are presented in Figure 1. At the end of weekly dosing, the mean daratumumab maximum Ctrough was 543.90 μg/mL for DARA-MD 1200 mg and 744.20 μg/mL for DARA-MD 1800 mg (Table 2). Notably, the PK concentrations of DARA-MD 1800 mg after the last (eighth) weekly dose were consistent with the last (seventh) weekly 16 mg/kg IV dose in GEN501 part 1. The profiles after the last weekly dose showed similar or greater Ctrough following subcutaneous dosing and similar variability and Cmax (Figure 2).

Daratumumab serum concentration-time profiles. Mean daratumumab serum concentration over time profiles after the first (A) and eighth (B) weekly doses of DARA-MD. The eighth weekly dose was the last weekly dose, which was administered on cycle 2 day 22; this profile ends with the predose concentration on cycle 3 day 1. Error bars represent ±1 standard deviation. *From study GEN501 part 2. †Cycle 2 day 22. ‡Predose on cycle 3 day 1. §From study GEN501 part 1.
Daratumumab serum concentration-time profiles. Mean daratumumab serum concentration over time profiles after the first (A) and eighth (B) weekly doses of DARA-MD. The eighth weekly dose was the last weekly dose, which was administered on cycle 2 day 22; this profile ends with the predose concentration on cycle 3 day 1. Error bars represent ±1 standard deviation. *From study GEN501 part 2. †Cycle 2 day 22. ‡Predose on cycle 3 day 1. §From study GEN501 part 1.
Maximum daratumumab Ctrough for DARA-MD 1200 mg, DARA-MD 1800 mg, and historical data for daratumumab 16 mg/kg IV
| Variable | Dose and route of administration | No. of patients | Mean daratumumab Ctrough on cycle 3 day 1, µg/mL | CV, % |
|---|---|---|---|---|
| DARA-MD (PAVO) | 1200 mg SC | 5 | 543.90 | 44 |
| DARA-MD (PAVO) | 1800 mg SC | 42 | 744.20 | 52 |
| Daratumumab (GEN501)2 | 16 mg/kg IV | 27 | 617.17 | 51 |
| Daratumumab (SIRIUS)16 | 16 mg/kg IV | 73 | 573.49 | 58 |
| Variable | Dose and route of administration | No. of patients | Mean daratumumab Ctrough on cycle 3 day 1, µg/mL | CV, % |
|---|---|---|---|---|
| DARA-MD (PAVO) | 1200 mg SC | 5 | 543.90 | 44 |
| DARA-MD (PAVO) | 1800 mg SC | 42 | 744.20 | 52 |
| Daratumumab (GEN501)2 | 16 mg/kg IV | 27 | 617.17 | 51 |
| Daratumumab (SIRIUS)16 | 16 mg/kg IV | 73 | 573.49 | 58 |
CV, coefficient of variation; SC, subcutaneously.
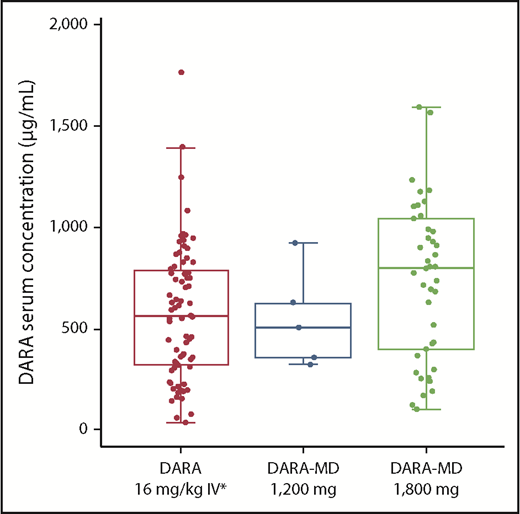
Daratumumab serum concentration at Ctrough on cycle 3 day 1 after DARA-MD or IV administration. Daratumumab Ctrough was determined at the end of weekly dosing as the predose concentration on cycle 3 day 1. *From studies GEN501 and SIRIUS.
Daratumumab serum concentration at Ctrough on cycle 3 day 1 after DARA-MD or IV administration. Daratumumab Ctrough was determined at the end of weekly dosing as the predose concentration on cycle 3 day 1. *From studies GEN501 and SIRIUS.
Anti-daratumumab antibodies were detected in 1 patient who received DARA-MD 1800 mg. The antibodies were neutralizing, transient, and did not appear to affect PK properties. At baseline (before treatment), 1 (12.5%) patient in the 1200-mg group and 4 (8.9%) patients in the 1800-mg group were positive for anti-rHuPH20 antibodies. During the study, 6 (13.3%) patients in the 1800-mg group and no patients in the 1200-mg group were positive for treatment-emergent anti-rHuPH20 antibodies (ie, patients who were negative at baseline and had at least 1 positive sample after the start of treatment) or treatment-boosted anti-rHuPH20 antibodies (ie, patients who were positive at baseline with at least 1 positive sample after the start of treatment with titers at least 2-fold compared with baseline). None of the anti-rHuPH20 antibodies was neutralizing, and there was no clear pattern for the timing or persistence of anti-rHuPH20 antibodies. The PK properties for patients who were positive for anti-rHuPH20 antibodies were similar to those of the overall study population. The presence of anti-rHuPH20 antibodies was not associated with an increased risk of IRRs or with an increased susceptibility for development of erythema or induration at the infusion site. Antitumor activity was observed in 3 of the 6 patients with anti-rHuPH20 antibodies, showing a confirmed response of PR or better.
Safety
At a median treatment duration of 2.6 months in the DARA-MD 1200-mg group and 5.4 months in the DARA-MD 1800-mg group, the safety profile of DARA-MD was tolerable. The most common (>25%) treatment-emergent AEs (TEAEs) in the 1200-mg group were thrombocytopenia (37.5%), upper respiratory tract infection (37.5%), insomnia (37.5%), and decreased appetite (37.5%) (Table 3). Grade 3 or 4 TEAEs occurring in >1 patient in the 1200-mg group included fatigue (25.0%) and hypertension (25.0%). In the 1800-mg group, the most common hematologic TEAEs included anemia (33.3%), thrombocytopenia (17.8%), lymphopenia (17.8%), and neutropenia (15.6%); the most common (>25%) nonhematologic TEAEs were upper respiratory tract infection, pyrexia, and diarrhea (26.7% each). Anemia was the most common grade ≥3 TEAE reported in the 1800-mg group, occurring in 7 (15.6%) patients. Other grade ≥3 TEAEs reported in >2 patients in the 1800-mg group included lymphopenia (11.1%), hypertension (8.9%), neutropenia (6.7%), and thrombocytopenia (6.7%). Serious TEAEs were reported in 4 (50.0%) patients in the 1200-mg group and in 14 (31.1%) patients in the 1800-mg group. One patient in the 1200-mg group and 3 patients in the 1800-mg group experienced serious TEAEs considered possibly drug related, including grade 2 pyrexia and grade 3 influenza B, lung infection influenza A, and dyspnea/musculoskeletal chest pain. With the exception of 1 TEAE-related death caused by aspiration pneumonia that occurred in the 1200-mg group, which was not considered to be related to treatment, no TEAE-related discontinuations were reported.
TEAEs
| TEAEs, n (%) | 1200 mg (n = 8) | 1800 mg (n = 45) | ||
|---|---|---|---|---|
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| >25% | >1 patient | >25% | >1 patient | |
| Hematologic | ||||
| Thrombocytopenia | 3 (37.5) | 1 (12.5) | 8 (17.8) | 3 (6.7) |
| Anemia | 2 (25.0) | 1 (12.5) | 15 (33.3) | 7 (15.6) |
| Neutropenia | 1 (12.5) | 1 (12.5) | 7 (15.6) | 3 (6.7) |
| Lymphopenia | 0 | 0 | 8 (17.8) | 5 (11.1) |
| Nonhematologic | ||||
| Upper respiratory tract infection | 3 (37.5) | 1 (12.5) | 12 (26.7) | 0 |
| Insomnia | 3 (37.5) | 0 | 5 (11.1) | 0 |
| Decreased appetite | 3 (37.5) | 0 | 3 (6.7) | 0 |
| Pyrexia | 2 (25.0) | 0 | 12 (26.7) | 0 |
| Diarrhea | 2 (25.0) | 0 | 12 (26.7) | 0 |
| Fatigue | 2 (25.0) | 2 (25.0) | 10 (22.2) | 1 (2.2) |
| Hypertension | 2 (25.0) | 2 (25.0) | 4 (8.9) | 4 (8.9) |
| Pneumonia | 1 (12.5) | 1 (12.5) | 4 (8.9) | 2 (4.4) |
| Hyponatremia | 0 | 0 | 2 (4.4) | 2 (4.4) |
| Respiratory syncytial virus infection | 0 | 0 | 2 (4.4) | 2 (4.4) |
| Device-related infection | 0 | 0 | 2 (4.4) | 2 (4.4) |
| TEAEs, n (%) | 1200 mg (n = 8) | 1800 mg (n = 45) | ||
|---|---|---|---|---|
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| >25% | >1 patient | >25% | >1 patient | |
| Hematologic | ||||
| Thrombocytopenia | 3 (37.5) | 1 (12.5) | 8 (17.8) | 3 (6.7) |
| Anemia | 2 (25.0) | 1 (12.5) | 15 (33.3) | 7 (15.6) |
| Neutropenia | 1 (12.5) | 1 (12.5) | 7 (15.6) | 3 (6.7) |
| Lymphopenia | 0 | 0 | 8 (17.8) | 5 (11.1) |
| Nonhematologic | ||||
| Upper respiratory tract infection | 3 (37.5) | 1 (12.5) | 12 (26.7) | 0 |
| Insomnia | 3 (37.5) | 0 | 5 (11.1) | 0 |
| Decreased appetite | 3 (37.5) | 0 | 3 (6.7) | 0 |
| Pyrexia | 2 (25.0) | 0 | 12 (26.7) | 0 |
| Diarrhea | 2 (25.0) | 0 | 12 (26.7) | 0 |
| Fatigue | 2 (25.0) | 2 (25.0) | 10 (22.2) | 1 (2.2) |
| Hypertension | 2 (25.0) | 2 (25.0) | 4 (8.9) | 4 (8.9) |
| Pneumonia | 1 (12.5) | 1 (12.5) | 4 (8.9) | 2 (4.4) |
| Hyponatremia | 0 | 0 | 2 (4.4) | 2 (4.4) |
| Respiratory syncytial virus infection | 0 | 0 | 2 (4.4) | 2 (4.4) |
| Device-related infection | 0 | 0 | 2 (4.4) | 2 (4.4) |
IRRs were reported for 1 (12.5%) patient who received DARA-MD 1200 mg and in 11 (24.4%) patients who received DARA-MD 1800 mg (Table 4). In the 1800-mg group, IRRs occurring in >1 patient included chills (8.9%), pyrexia (6.7%), hypotension, pruritus, and paresthesia (4.4% each). IRRs were generally grade 1/2 in severity; grade 3 IRRs were reported in 1 patient in the 1200-mg group (grade 3 dyspnea in combination with grade 2 chills and grade 1 noncardiac chest pain) and in 1 patient in the 1800-mg group (hypertension). The IRR of grade 3 hypertension (156/101 mm Hg) was reported concurrent with other grade 1/2 IRRs (ie, flushing, sinus pain, oropharyngeal pain, pruritus) in this patient. No grade 4 IRRs were reported. In the 1200-mg group, the single patient with IRRs experienced the events with the first infusion. Of the 11 patients in the 1800-mg group who experienced IRRs, 10 experienced an IRR during the first infusion and 2 experienced an IRR in the third or later infusions. One patient developed grade 2 hypotension after cycle 1 day 22 administration that was managed with infusion of a saline solution, and another patient reported on cycle 6 day 1, cycle 12 day 1, and cycle 18 day 1 variable grade 1/2 IRRs (tickling in the throat, cold sweat, hyperhidrosis, paresthesia, and throat irritation) that responded well to supportive therapy.
All grade IRRs
| IRRs, n (%) | 1200 mg (n = 8) | 1800 mg (n = 45) |
|---|---|---|
| Chills | 1 (12.5) | 4 (8.9) |
| Pyrexia | 0 | 3 (6.7) |
| Hypotension | 0 | 2 (4.4) |
| Pruritus | 0 | 2 (4.4) |
| Paresthesia | 0 | 2 (4.4) |
| Noncardiac chest pain | 1 (12.5) | 0 |
| Dyspnea* | 1 (12.5) | 0 |
| Oropharyngeal pain | 0 | 1 (2.2) |
| Pharyngeal paresthesia | 0 | 1 (2.2) |
| Sinus pain | 0 | 1 (2.2) |
| Throat irritation | 0 | 1 (2.2) |
| Wheezing | 0 | 1 (2.2) |
| Hypertension* | 0 | 1 (2.2) |
| Flushing | 0 | 1 (2.2) |
| Nausea | 0 | 1 (2.2) |
| Tongue edema | 0 | 1 (2.2) |
| Vomiting | 0 | 1 (2.2) |
| Cold sweat | 0 | 1 (2.2) |
| Hyperhidrosis | 0 | 1 (2.2) |
| Rash | 0 | 1 (2.2) |
| IRRs, n (%) | 1200 mg (n = 8) | 1800 mg (n = 45) |
|---|---|---|
| Chills | 1 (12.5) | 4 (8.9) |
| Pyrexia | 0 | 3 (6.7) |
| Hypotension | 0 | 2 (4.4) |
| Pruritus | 0 | 2 (4.4) |
| Paresthesia | 0 | 2 (4.4) |
| Noncardiac chest pain | 1 (12.5) | 0 |
| Dyspnea* | 1 (12.5) | 0 |
| Oropharyngeal pain | 0 | 1 (2.2) |
| Pharyngeal paresthesia | 0 | 1 (2.2) |
| Sinus pain | 0 | 1 (2.2) |
| Throat irritation | 0 | 1 (2.2) |
| Wheezing | 0 | 1 (2.2) |
| Hypertension* | 0 | 1 (2.2) |
| Flushing | 0 | 1 (2.2) |
| Nausea | 0 | 1 (2.2) |
| Tongue edema | 0 | 1 (2.2) |
| Vomiting | 0 | 1 (2.2) |
| Cold sweat | 0 | 1 (2.2) |
| Hyperhidrosis | 0 | 1 (2.2) |
| Rash | 0 | 1 (2.2) |
Grade 3 IRR; all other IRRs were grades 1 to 2.
IRRs, if present, mostly developed during or within 6 hours after the start of the subcutaneous infusion. In both dose groups, 12 patients experienced a total of 32 IRRs. The exact onset time was reported for 29 IRRs and was not available for 3 IRRs reported on day 1 of treatment. Based on the start time of infusion and IRR onset time, 31.3% of IRRs (3 patients) were reported within the first 60 minutes from the start of infusion, 3.1% of IRRs (1 patient) within 60 to 120 minutes from the start of infusion, 9.4% of IRRs (1 patient) within 120 to 180 minutes from the start of infusion, 12.5% of IRRs (1 patient) within 180 to 240 minutes from the start of infusion, 18.8% of IRRs (3 patients) within 240 to 300 minutes from the start of infusion, and 15.6% of IRRs (3 patients) within 300 to 360 minutes from the start of infusion. Treatment modification (infusion interruption) due to IRRs was required in 2 (4.4%) patients on cycle 1 day 1 and in 1 (2.2%) patient on cycle 1 day 21, all of whom received the full dose of DARA-MD 1800 mg, despite infusion interruption. Overall IRRs, if present, could be well controlled with supportive therapy, including antihistaminic, corticosteroids, or bronchodilator therapy.
Subcutaneous delivery of daratumumab in the periumbilical area was well tolerated despite the administration volume of 60 or 90 mL. Infusion site TEAEs were reported only in the 1800-mg group and included pain at the infusion/injection site (3 [6.7%] patients), infusion/injection site erythema (3 [6.7%] patients), induration (1 [2.2%] patient), and paresthesia (1 [2.2%] patient). All infusion site TEAEs were grade 1 in severity and resolved without intervention. Infusion site reactions of erythema or induration during treatment (based on objective assessment of presence/absence and measurement, as required per protocol) were observed for patients in both dose groups. In the 1200-mg group, erythema and induration were reported in 5 (62.5%) and 4 (50.0%) patients, respectively. In the 1800-mg group, erythema occurred in 13 (28.9%) patients, and induration was reported in 10 (22.2%) patients. All measurable local infusion site reactions resolved within 1 to 2 hours after the infusion ended.
Efficacy
At a median follow-up of 5.2 months, an ORR of 25.0% was achieved in the 1200-mg group, which included 2 PRs, with a median duration of response of 6.4 months (Figure 3A). For patients in the 1200-mg group, median time to first response was 3.4 months. With a median follow-up of 8.3 months, patients receiving DARA-MD 1800 mg achieved an ORR of 42.2%, including stringent CR in 4 (8.9%) patients, very good PR (VGPR) in 5 (11.1%) patients, and PR in 10 (22.2%) patients. The median time to first response among patients in the 1800-mg group was 1.0 month, and the median duration of response was 14.2 months. Among responders who received DARA-MD 1800 mg, many responses deepened over time. Among the 10 patients with an initial PR, 3 went on to achieve stringent CR and 4 to achieve VGPR with further treatment. In addition, an initial response of VGPR in 1 patient receiving DARA-MD 1800 mg continued to deepen to stringent CR (Figure 3B).
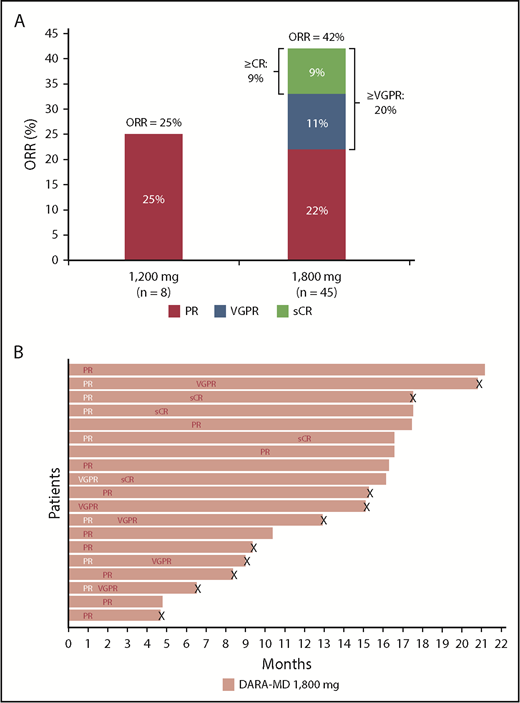
Responses in the DARA-MD 1800 mg group. Summary of responses (A) and swim lane plot of responders in the DARA-MD 1800 mg group (B). Responses were evaluated in the overall study population.
Responses in the DARA-MD 1800 mg group. Summary of responses (A) and swim lane plot of responders in the DARA-MD 1800 mg group (B). Responses were evaluated in the overall study population.
Discussion
These findings show that daratumumab could be safely combined with rHuPH20 as a first-generation formulation that allowed subcutaneous administration of a therapeutic 1800-mg dose over 30 minutes. DARA-MD was well tolerated, with an AE profile comparable to IV administration and a lower IRR rate, which mainly occurred with the first administration and mostly developed during or within 6 hours after the start of the infusion. Durable responses were reported in the 1800-mg group consistent with IV administration in a similar patient population.2 In a pooled analysis of the daratumumab IV monotherapy studies GEN501 and SIRIUS (N = 148), patients with heavily pretreated RRMM (median of 5 [2-14] previous lines of therapy) achieved an ORR of 31.3%, including 13.5% VGPR or better,18 compared with the 42.2% ORR and 20.0% VGPR or better rates observed with DARA-MD. In addition, PK concentrations achieved with the DARA-MD 1800-mg dose were consistent with daratumumab 16 mg/kg IV in patients with RRMM, with a lower peak concentration reached after the initial dose of DARA-MD and a similar or greater maximum Ctrough at the end of weekly dosing (before the cycle 3 day 1 dose). The variability in daratumumab concentrations was consistent despite the fact that IV dosing is administered based on individual patient weight, whereas the subcutaneous dose is fixed.19,20 Population PK analysis and simulations for daratumumab indicated that either weight-based dosing or fixed-dosing protocols are reasonable approaches for drug administration, and published literature has provided strong support for using fixed dosing of monoclonal antibodies in clinical trials in adults.19-21 Therefore, a fixed-dosing approach was selected to simplify dose administration.
Similar to studies of daratumumab IV, there was a low incidence of anti-daratumumab antibodies (1.3%), indicating a low risk of daratumumab immunogenicity with subcutaneous infusion (data on file). Six patients (13.3%) were positive for treatment-emergent or treatment-induced anti-rHuPH20 antibodies, similar to the rate reported for other rHuPH20-containing treatments (subcutaneous rituximab [9%], trastuzumab [20%], and immune globulin infusion HYQVIA [18%]).11,12,22 Because rHuPH20 is a recombinantly expressed version of human hyaluronidase PH20, there is a potential for preexisting antibodies to endogenous proteins capable of binding to rHuPH20. Five patients (9.4%), including 1 patient in the 1200-mg cohort, had a rHuPH20-positive sample at baseline. This rate is in the range of the previously reported prevalence of baseline rHuPH20-binding antibodies (3%-12%).23 None of the samples positive for rHuPH20 antibodies was neutralizing, and there was no clear pattern for the timing or persistence of the presence of anti-rHuPH20 antibodies.
Because IV administration requires dedicated facilities, equipment, and personnel, and is associated with long infusion times and risk of complications and IRRs,24-26 subcutaneous administration offers a preferable alternative. Coformulation with rHuPH20 helps overcome some of the challenges of subcutaneous dosing of large volumes by enabling drug dispersion and absorption at the administration site.26 When combined with rHuPH20, trastuzumab administered subcutaneously greatly reduced infusion times in patients with breast cancer,12,27 while maintaining efficacy and tolerability comparable to IV administration.27 Studies of subcutaneously administered rituximab in patients with lymphoma reported similar findings.25,28 Subsequently, subcutaneous formulations of rHuPH20 in combination with trastuzumab (Herceptin SC) or rituximab (MabThera SC) received approval in Europe and the United States (Herceptin Hylecta and Rituxan Hycela, respectively).11-14
The findings presented here reveal that daratumumab can be safely coadministered with rHuPH20, showing acceptable tolerability with drastically reduced infusion time. The safety profile of DARA-MD was consistent with IV daratumumab, with no new safety signals. Importantly, DARA-MD was associated with a low incidence of IRRs, occurring in only 1 (13%) patient in the 1200-mg group and 11 (24%) patients in the 1800-mg dose group. IRRs were generally grade 1/2 in severity, with grade 3 IRRs reported in 2 (4%) patients and no grade 4 IRRs reported. Because IRRs occur in ∼50% of patients treated with daratumumab IV, and this finding therefore represents a promising reduction in the rate of IRRs associated with subcutaneous daratumumab. Furthermore, abdominal wall injections of DARA-MD were well tolerated. Although objectively measured erythema and induration were reported in some patients, these events resolved quickly (within 1 to 2 hours after infusion).
Responses to DARA-MD were observed across both dose groups, with deeper responses reported in the 1800-mg cohort. The efficacy of DARA-MD 1800 mg was consistent with that of daratumumab IV 16 mg/kg in a similar patient population,2 with responses to treatment deepening with longer follow-up. Investigations of the PK profile of daratumumab IV 16 mg/kg have shown that this dose achieves and maintains concentrations consistent with target saturation, with a dosing schedule that is initially more intensive and then becomes less frequent to overcome the period of initial higher clearance.29 This same dosing schedule was used for DARA-MD, and a similar PK profile was observed. By achieving a similar or greater serum concentration at trough, with a slower absorption into systemic circulation, DARA-MD allows for consistent efficacy compared with the IV formulation, with a drastically shorter infusion time and a reduction in the IRR rate.
Based on the positive safety and efficacy findings with DARA-MD, part 2 of PAVO was initiated to investigate the safety and efficacy of a premixed concentrated coformulation of daratumumab 1800 mg with rHuPH20, administered manually via handheld syringe (DARA-SC 1800 mg). This formulation contains a higher concentration of daratumumab in a lower injection volume, enabling dosing in 3 to 5 minutes via subcutaneous injection into the abdomen.
Taken together, these findings show that subcutaneous administration of daratumumab is feasible in patients with MM. The 1800-mg dose of DARA-MD achieves PK concentrations similar to or greater than those after IV infusion of daratumumab 16 mg/kg, achieves deep and durable responses in patients with MM, and has an acceptable safety profile with a low rate of IRRs.
The data-sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated in this study and their families, as well as the study coinvestigators, research nurses, and coordinators at each of the clinical sites.
This study was sponsored by Janssen Research & Development, LLC. Medical writing and editorial support were provided by Kristin Runkle of MedErgy and were funded by Janssen Global Services, LLC.
Study data were collected by the investigators and their research teams. Final data analysis and verification of accuracy were conducted by Janssen. The investigators were not restricted by confidentiality agreements and had full accessibility to all the data.
Authorship
Contribution: S.Z.U., H.N., P.H., T.M., and J.S.-M. contributed to the study design, data acquisition, and data interpretation or analysis; M.-V.M., N.W.C.J.v.d.D., A.C., P.M., A.O., and T.P. contributed to the acquisition of data, data interpretation, and data analysis; J.L.K. and L.B. contributed to the acquisition of data; P.L.C. contributed to the study design and data interpretation or analysis; M.L. contributed to the data analysis and interpretation; K.L. contributed to the study design; and all authors drafted and reviewed the manuscript, approved the final version, decided to publish this report, and vouch for data accuracy and completeness.
Conflict-of-interest disclosure: S.Z.U. consulted for AbbVie, GlaxoSmithKline, Celgene, Amgen/Onyx, Takeda/Millennium, Sanofi, Seattle Genetics, Skyline, Merck, and Janssen; received research funding from Celgene, Amgen/Onyx, Takeda/Millennium, Sanofi, Seattle Genetics, Skyline, Merck, Janssen, Array BioPharma, and Pharmacyclics; served on speakers bureaus for Celgene, Amgen, Janssen, Sanofi, and Takeda; and received travel expenses from Janssen, Celgene, Amgen, and Takeda. M.-V.M. received honoraria from and consulted for Celgene, Janssen, Takeda, and Amgen. N.W.C.J.v.d.D. received research support from Janssen Pharmaceuticals, Amgen, Celgene, Novartis, and Bristol-Myers Squibb; and served on advisory boards for Janssen Pharmaceuticals, Amgen, Celgene, Bristol-Myers Squibb, Novartis, Bayer, Takeda, and Servier. A.C. consulted for Amgen, Array BioPharma, Celgene, Janssen, Millennium, Takeda, and Novartis; and received research funding from Amgen, Array BioPharma, Celgene, Janssen, Millennium, Takeda, Novartis, and Pharmacyclics. J.L.K. consulted for AbbVie, Roche, Takeda, Janssen, and Pharmacyclics; and received research funding from Amgen and Novartis. P.M. consulted for and received honoraria from Celgene, Takeda, and Janssen. A.O. consulted for and received honoraria from Amgen, Takeda, and Janssen; and served on speakers bureaus for Amgen and Janssen. T.P. received research support from Janssen Pharmaceuticals; and served on advisory boards for Janssen Pharmaceuticals, Celgene, Takeda, and Behring. L.B. consulted for and received honoraria from Takeda, Celgene, Janssen, and Amgen; and received travel expenses from Janssen, Celgene, and Amgen. P.H., T.M., P.L.C., M.L., and K.L. are employees of Janssen. P.H., T.M., P.L.C., and M.L. hold stock in Johnson & Johnson. J.S.-M. consulted for Amgen, Bristol-Myers Squibb, Celgene, Janssen, Merck Sharp & Dohme, Novartis, Takeda, Sanofi, and Roche. H.N. declares no competing financial interests.
Correspondence: Saad Z. Usmani, Department of Hematologic Oncology & Blood Disorders, Levine Cancer Institute/Atrium Health, 1021 Morehead Medical Dr, Charlotte, NC 28204; e-mail: saad.usmani@atriumhealth.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal