

TO THE EDITOR:
Chronic neutrophilic leukemia (CNL) is a rare myeloproliferative neoplasm characterized by sustained mature neutrophil proliferation, bone marrow (BM) granulocytic hyperplasia, and splenomegaly.1 For a long time, the diagnosis of CNL has been based on the exclusion of genetic abnormalities that are known to occur in other myeloproliferative neoplasms. In 2013, the discovery of recurrent mutations in the CSF3R gene in the majority of patients with CNL has revolutionized the understanding of the pathogenesis of this disease and provided a biomarker for CNL diagnosis.2 Consequently, the World Health Organization revised the diagnostic criteria for CNL in 2016 to include CSF3R mutations (T618I or other activating CSF3R mutation).3
The CSF3R gene encodes the colony-stimulating factor 3 receptor (also known as granulocyte colony–stimulating factor [G-CSF] receptor) which is critical for differentiation and proliferation of neutrophils.4 The structure of the receptor comprises an N-terminal extracellular part involved in G-CSF binding and a C-terminal cytoplasmic domain involved in the activation of downstream kinase signaling pathways.5 The most common mutation in CNL is a threonine-to-isoleucine substitution in the membrane-proximal extracellular domain of the receptor, at codon 618 (T618I, previously referred to as T595I using the historical numbering that excludes the 23-amino-acid signal peptide). The T618I mutation has been shown to increase receptor dimerization and confer ligand-independent activation with subsequent neutrophilic expansion primarily through increased JAK-STAT signaling.6
Here, we report for the first time an inherited transmission of the CSF3R T618I mutational hotspot in a pedigree with multiple diagnoses of CNL (Figure 1A). The biological samples were collected after informed consent, in accordance with the Declaration of Helsinki.
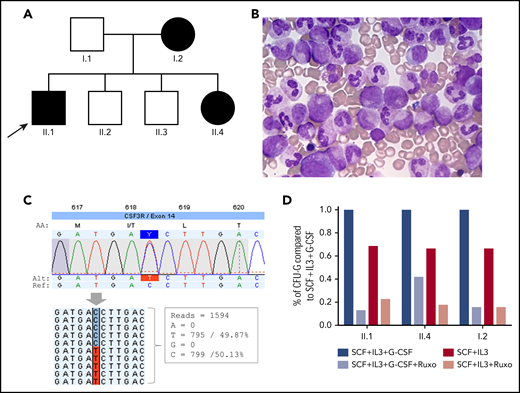
Germline CSF3R c.1853C>T:p.T618I mutation. (A) Family tree. Arrow indicates the proband. Individuals with CNL and germline T618I mutation are marked in black. (B) BM aspirate in individual II.1 showing granulocytic hyperplasia. (C) Schematic of the CSF3R T618I mutation identified by HTS. Reads carrying the wild-type nucleotide (n = 799) and the variant nucleotide (n = 795) are shown (screenshot from JSI Medical System software). (D) Clonogenic assay. CD34+ progenitors (n = 1000) were seeded in methylcellulose in SCF + IL-3 ± G-CSF ± ruxolitinib; CFU-G colonies were counted 14 days later. The results are the means ± SD of 1 representative experiment performed in triplicate. IL-3, interleukin-3; SCF, stem cell factor.
Germline CSF3R c.1853C>T:p.T618I mutation. (A) Family tree. Arrow indicates the proband. Individuals with CNL and germline T618I mutation are marked in black. (B) BM aspirate in individual II.1 showing granulocytic hyperplasia. (C) Schematic of the CSF3R T618I mutation identified by HTS. Reads carrying the wild-type nucleotide (n = 799) and the variant nucleotide (n = 795) are shown (screenshot from JSI Medical System software). (D) Clonogenic assay. CD34+ progenitors (n = 1000) were seeded in methylcellulose in SCF + IL-3 ± G-CSF ± ruxolitinib; CFU-G colonies were counted 14 days later. The results are the means ± SD of 1 representative experiment performed in triplicate. IL-3, interleukin-3; SCF, stem cell factor.
The proband (II.1) is an 18-year-old man referred to our center for abdominal pain, splenomegaly, and unexplained leukocytosis. Personal and familial history was unremarkable. The full blood count showed: white blood cell (WBC) count 30 × 109/L (including 87% mature neutrophils), hemoglobin 13 g/dL, and platelet count 263 × 109/L. Serial blood counts showed persistent leukocytosis (range, 21-38 × 109/L). The BM aspirate was hypercellular with granulocytic hyperplasia and no dysplasia (Figure 1B). Immunophenotyping in peripheral blood (PB) showed a normal repartition of monocyte subsets and no abnormal cells. Cytogenetic analyses were normal. Molecular screening for BCR-ABL1, JAK2, CALR and MPL was negative. However, targeted high-throughput sequencing (HTS) (supplemental Methods, available on the Blood Web site) revealed a heterozygous T618I mutation in exon 14 of CSF3R (Figure 1C). Mutations in other genes were absent.
Four months later, the observation of splenomegaly and leukocytosis was incidentally made in his sister (II.4; age, 9 years) during laboratory tests for exploration of an atopic background. Her blood count showed 24 × 109/L WBC including 86% mature neutrophils. HTS in PB identified the heterozygous CSF3R T618I mutation (variant allele frequency [VAF], 50%). Another heterozygous variant of uncertain significance (VUS) in NRAS (c.54_55delAC:p.L19Dfs*12; VAF, 49%) was also identified (supplemental Figure 1). Considering the previous diagnosis of CNL in the pedigree, a skin biopsy with fibroblast culture was performed in patient II.4. HTS on germline DNA extracted from fibroblasts finally demonstrated the constitutional origin of the CSF3R mutation (and the NRAS variant with the same VAFs). Blood counts performed in individuals I.1, II.2, and II.3 were normal, but the mother (I.2; age, 37 years) had leukocytosis (WBC, 22 × 109/L with 84% mature neutrophils) and splenomegaly. Molecular analyses revealed the presence of the CSF3R T618I mutation (and the NRAS VUS). She had no significant medical history but had displayed a stable and unexplained leukocytosis since age 12.
Clonogenic assay was performed as previously described7 in individuals I.2, II.1, and II.4. Briefly, CD34+ progenitors were purified from PB of patients and cultured in methylcellulose in the presence of stem cell factor (25 ng/mL) and interleukin-3 (10 ng/mL) with or without G-CSF (20 ng/mL). In the absence of G-CSF, we found a major spontaneous growth of granulocyte colony-forming unit (CFU-G) progenitors (∼65%) in patient cells carrying the CSF3R T618I mutation showing that progenitors can grow independently of G-CSF by contrast to normal CD34+ cells (not shown).7 Moreover, we found that both normal and spontaneous growth of CFU-G were sensitive to JAK inhibition using the JAK1/2 inhibitor ruxolitinib (Figure 1D).
Finally, considering these results and previous reports8 and because the proband II.1 has experienced new episodes of abdominal pain related to splenomegaly, it was decided to start ruxolitinib 10 mg ×2/d. After 4 months, the full blood count showed: WBC 16 × 109/L (with 76% mature neutrophils), hemoglobin concentration 14 g/dL, and platelet count 288 × 109/L. Ruxolitinib dosage was increased to 15 mg ×2/d. Unfortunately, the patient did not attend the next medical consultations and follow-up was not performed.
To date, a single case in a child carrying a germline CSF3R T618I mutation was reported in 2016. The patient was an 11-year-old girl with persistent leukocytosis (range, 24-37 × 109/L) and splenomegaly. However, neither parent carried the mutation, suggesting it had arisen as a result of either genetic mosaicism or a spontaneous mutation in utero.9 Previously, the germline CSF3R T640N mutation (referred as T617N in the old nomenclature) has been described in a large pedigree with multiple cases of chronic neutrophilia.7 The CSF3R T640N mutation is located in the transmembrane domain of the protein and also causes ligand-independent activation with increased receptor dimerization.7,10 The mutation was found in 12 of 16 members through 3 generations with complete penetrance. All affected subjects were diagnosed with splenomegaly and leukocytosis (range, 15-33 × 109/L). Together, these observations raise the possibility of a germline disorder in individuals diagnosed with CNL. In clinical practice, control of a germline sample is not routinely performed at the time of diagnosis. Thus, it cannot be excluded that this situation is currently underestimated.
It is remarkable that all affected members in the present family had normal development without evidence of neutrophil-related disorder, such as infections or inflammation. The older individual was 39 years of age at the time of this report and had stable leukocytosis without any sign of disease evolution. Our report demonstrates that the effect of the CSF3R T618I mutation is restricted to the granulocytic compartment and is sufficient to induce proliferation, in line with mouse BM transplant models.5
The risk of disease evolution remains the main preoccupation because blast transformation has been reported to occur in a significant proportion (∼20%) of patients with CNL.1 Patients often carry additional clonal aberration in the blastic phase, and some events, including ASXL1 and SETBP1 mutations, have been associated with an increased risk of evolution.11,12 This provides strong evidence that CNL progression is a multistep process probably requiring additional genetic lesions. Thus, a reasonable approach in germline CSF3R-mutated carriers could be to perform complete blood count monitoring, with molecular analysis to detect clonal evolution and BM biopsy limited to those with progressive cytopenia, increased leukocytosis, or abnormal blood cells. In all affected subjects in the present pedigree, no additional mutation was found in CSF3R. A germline NRAS heterozygous frameshift VUS was found in I.2 and II.4 but not in II.1. Although this frameshift variant is supposed to lead to the premature truncation of the protein, all oncogenic mutations in the RAS gene family members reported to date include point mutations (more rarely, in-frame indel) involving highly conserved codons leading to constitutive activation of the protein. No significant difference in biological findings, including WBC count or progenitor growth, was observed in individuals who carried this additional variant compared with the proband who did not.
In conclusion, we report the first family with CNL resulting from the constitutional CSF3R T618I mutation. This suggests that the germline CSF3R status should be sought in further studies to determine the true prevalence of this disorder.
All sequence data, BAM files, VCF files, are available on demand via the corresponding author.
The online version of this article contains a data supplement.
Acknowledgment
The authors thank Christophe Roumier for handling, conditioning, and storing patient samples (Lille Hospital Tumor Bank, certification NF 96900-2014/65453-1).
Authorship
Contribution: N.D., A.M.-R., L.F., O.N., and C.P. performed molecular analyses; T.B. performed morphological examinations and flow cytometry; G.H. performed fibroblast culture; C.W., I.P., and S.d.B. performed clonogenic assays; C.R., B.N., and B.Q. provided samples and clinical data; and N.D., C.W., I.P., and C.P. wrote the manuscript, which was approved by all the authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nicolas Duployez, Laboratory of Hematology, Biology and Pathology Center, Lille University Hospital, Professor J. Leclercq Blvd, 59037 Lille Cedex, France; e-mail: nicolas.duployez@chru-lille.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal