


Abstract
Recurrent mutations in calreticulin are present in ∼20% of patients with myeloproliferative neoplasms (MPNs). Since its discovery in 2013, we now have a more precise understanding of how mutant CALR, an endoplasmic reticulum chaperone protein, activates the JAK/STAT signaling pathway via a pathogenic binding interaction with the thrombopoietin receptor MPL to induce MPNs. In this Spotlight article, we review the current understanding of the biology underpinning mutant CALR-driven MPNs, discuss clinical implications, and highlight future therapeutic approaches.
Medscape Continuing Medical Education online
![]()
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 2332.
Disclosures
Associate Editor John Crispino received grants for laboratory research from Forma Therapeutics and Scholar Rock and served as an advisor or consultant for the MPN Research Foundation. Author Gabriela S. Hobbs served as advisor or consultant for Agios Pharmaceuticals, Celgene Corporation, Incyte Corporation, and Jazz Pharmaceuticals, Inc. and received grants for clinical research from Bayer HealthCare Pharmaceuticals, Incyte Corporation, and Merck & Co., Inc. Author Ann Mullally served as advisor or consultant for CTI BioPharma and served as a speaker or a member of a speakers bureau for Blueprint Medicines, Incyte Corporation, and Roche. CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC and author Joan How declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe mechanistic and biochemical data regarding mutant CALR's role as a driver mutation in myeloproliferative neoplasms (MPNs), according to a review
Determine clinical data regarding mutant CALR's role as a driver mutation in MPNs, according to a review
Identify current treatment of MPNs with mutant CALR and therapeutic targeting of CALR, according to a review
Release date: December 19, 2019; Expiration date: December 19, 2020
Introduction
Molecular understanding of myeloproliferative neoplasm (MPN) pathogenesis was transformed with the finding that driver mutations in JAK2 occur in essentially all cases of polycythemia vera and ∼50% of essential thrombocythemia (ET) and primary myelofibrosis (PMF).1-4 Soon thereafter, activating mutations in MPL, the gene encoding the thrombopoietin receptor were identified in ∼3% to 5% of ET patients and 5% to 10% of PMF patients.5-8 The remaining molecular gap in MPN was filled in 2013 with the discovery that mutations in the gene encoding calreticulin (CALR) occurred in the majority of non-JAK2/MPL–mutated ET and PMF patients.9,10 The purpose of this Spotlight is to summarize the mechanistic, biochemical, and clinical data published on mutant CALR’s role as a driver mutation in MPN and highlight how these findings can inform directions for future therapeutic approaches.
Calreticulin structure and function
In 2013, 2 studies used whole-exome sequencing to identify the presence of recurrent mutations in CALR in 70% to 80% of ET and PMF patients without a JAK2 or MPL mutation.9,10 These mutations consist of insertions and/or deletions in exon 9, resulting in the generation of a novel mutant-specific positively charged amino acid sequence in the C terminus.9,10 The 2 most common mutations are a 52-bp deletion (L367fs*46) and a 5-bp insertion (K385fs*47), initially termed type 1 and type 2 mutations, respectively.9 Type 1 mutations eliminate all of the negatively charged amino acids in the CALR C terminus, whereas type 2 mutations eliminate approximately half of the negatively charged amino acids.11 All other mutations can be categorized as type 1 like or type 2 like, depending on the extent of amino acid deletion. Since the initial discovery of CALR mutations in MPN, >50 mutations have been described; however type 1 and type 2 mutations make up 80% of all mutations. Importantly, all CALR mutations have a common effect in creating a +1-bp frameshift in exon 9, resulting in the generation of a mutant-specific C terminus that is shared across all CALR-mutant MPNs, consistent with a gain of function.
CALR is an endoplasmic reticulum (ER) chaperone protein with functions in protein folding quality control and calcium homeostasis.12 Its protein structure has 3 domains: an amino domain, which is essential for chaperone function via its lectin binding sites and contains an ER signal peptide sequence; a proline-rich P domain, which binds to calcium and has a chaperone lectin binding site; and a carboxyl domain, which also functions in calcium binding and includes an ER-retention signal (KDEL motif).13 CALR mutations result in loss of the KDEL motif and the generation of a novel positively charged C terminus. Mutations in CALR are typically heterozygous, although homozygous mutations can occur.14
Mechanism of mutant CALR-induced oncogenesis
Mutations in CALR are present in the long-term hematopoietic stem cell compartment of MPN patients, where they can be found as the sole mutation, consistent with a disease-initiating role for mutant CALR in MPN.10 Retroviral, transgenic, and knock-in mutant CALR mouse models all engender an MPN phenotype that closely recapitulates human MPN, further supporting a disease-initiating role for mutant CALR.15-19 Furthermore, the ET-like phenotype in CALRdel52 knock-in mice is transplantable, indicating a hematopoietic stem cell–intrinsic effect of mutant CALR.17,19
It was not immediately apparent how recurrent mutations in CALR induce disease. Subsequent investigation from several groups has since established the biologic requirements for mutant CALR-induced oncogenesis, which include expression of MPL and its N-glycosylation sites,15,18,20-22 the mutant-specific C terminus of mutant CALR and, in particular, its positive electrostatic charge,15,18 a physical interaction between mutant CALR and MPL,18,20 and the lectin-dependent function of mutant CALR.21,23
Figure 1 illustrates the current understanding of the mechanism of mutant CALR-induced oncogenesis. Mutant CALR entry into the ER secretory pathway is required for MPL activation, and loss of mutant CALR’s signal peptide abrogates STAT5 transcriptional activity.24 Once outside the ER, mutant CALR forms stable complexes with preprocessed forms of MPL containing immature N-glycosylation sites21,24 ; this interaction is dependent on mutant CALR’s lectin-binding domain.23,24 The bound mutant CALR-MPL complexes are present in the Golgi apparatus and are then trafficked to the cell surface.24 The interaction with mutant CALR allows thrombopoietin-independent dimerization of MPL’s cytosolic tails, with cell surface localization leading to full receptor activation.24 Interestingly, a recent study has shown that the mutated C terminus allows the formation of mutant CALR homo-multimers and has suggested it is this homo-multimer structure that allows activation of MPL.25 The net result of the mutant CALR-MPL pathogenic binding interaction is ligand-independent MPL-JAK/STAT signaling activation resulting in clonal expansion of long-term hematopoietic stem cells and megakaryocytes. Activation of the unfolded protein response has been demonstrated at the transcriptional level in CALR-mutant MPN, in addition to upregulation of the NF-κB pathway.26,27 Interestingly, mutant CALR has been shown to have altered cellular localization because of loss of its C-terminal KDEL sequence, resulting in new protein-binding interactions, including in the nucleus.28
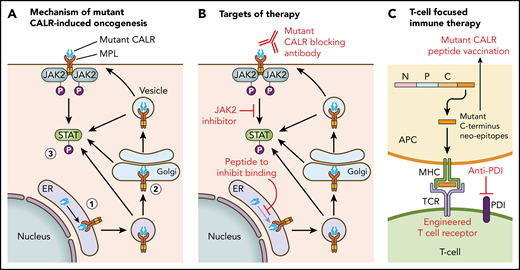
Mechanism of mutant CALR-induced MPN and approaches for therapeutic targeting. (A) A pathogenic binding interaction between MPL and mutant CALR leads to activated MPL-JAK/STAT signaling. 1. Mutant CALR traffics through the ER to bind to immature MPL. 2. Stabilized mutant calreticulin-MPL complex traffics to the cell surface. 3. Mutant CALR induces MPL-JAK/STAT signaling pathway activation. (B) Potential nodes for therapeutic intervention in mutant-CALR–driven MPN. (C) Strategies to induce T-cell–directed immune therapy against mutant-CALR–driven MPN. APC, antigen-presenting cell; MPL, major histocompatibility complex; TCR, T-cell receptor.
Mechanism of mutant CALR-induced MPN and approaches for therapeutic targeting. (A) A pathogenic binding interaction between MPL and mutant CALR leads to activated MPL-JAK/STAT signaling. 1. Mutant CALR traffics through the ER to bind to immature MPL. 2. Stabilized mutant calreticulin-MPL complex traffics to the cell surface. 3. Mutant CALR induces MPL-JAK/STAT signaling pathway activation. (B) Potential nodes for therapeutic intervention in mutant-CALR–driven MPN. (C) Strategies to induce T-cell–directed immune therapy against mutant-CALR–driven MPN. APC, antigen-presenting cell; MPL, major histocompatibility complex; TCR, T-cell receptor.
Intriguingly, mutant CALR protein is detectable in the plasma of CALRdel52 knock-in mice19 and CALR-mutant MPN patients,29 findings that build on earlier work in experimental model systems indicating that mutant CALR is secreted.30,31 Although, under experimental conditions, high concentrations of recombinant mutant CALR protein were shown to stimulate MPL when it is bound to mutant CALR on the cell surface, it is unclear how relevant this finding is to MPL activation in MPN patients.29 Importantly, cell surface expression of mutant CALR, albeit at a low level, was recently demonstrated on CALR-mutant CD34+ cells in a small number of MPN patients.24 Of note, it was previously demonstrated that cell surface mutant CALR does not induce MPN through inhibition of phagocytosis.32
Clinical significance of mutant calreticulin in MPNs
CALR-mutant ET and PMF patients have distinct clinical characteristics and outcomes from JAK2- and MPL-mutant patients (Table 1). Compared with JAK2-mutant ET patients, CALR-mutant ET patients tend to be younger with lower hemoglobin, decreased leukocytosis, and higher platelet counts, and there is a higher male preponderance.7,9,10,33,34 CALR-mutant ET patients have a higher male predominance compared with MPL-mutant ET patients but otherwise display similar laboratory values, including elevated platelet counts, consistent with a shared phenotype of preferential megakaryocytic expansion.33,34 Similarly, CALR-mutant PMF patients are younger, with a lower incidence of anemia and leukocytosis and higher platelet counts.8,34-36 Within PMF, CALR-mutant patients have lower International Prognostic Scoring System and Dynamic International Prognostic Scoring System (DIPSS) scores. Absence of a CALR mutation has been into incorporated into more recent PMF prognostic scoring systems (eg, Myelofibrosis Secondary to PV and ET-Prognostic Model [MYSEC-PM],37 Mutation-enhanced International Prognostic Scoring System for transplant-eligible patients [MIPSS70],38 and Genetically Inspired Prognostic Scoring System [GIPSS]39 ).
Summary of clinical features and outcomes of CALR-mutant ET and MF
| CALR vs JAK2 | CALR vs MPL | CALR vs triple negative | |
|---|---|---|---|
| ET | |||
| Clinical | Younger, male predominance, lower WBC count, lower Hg/Hct, higher platelets | Male predominance, otherwise similar | Male predominance, higher platelets |
| Thrombosis | Decreased risk | Decreased risk | Similar |
| Post-ET MF | Similar to increased | Similar | Similar |
| Overall prognosis | Similar | Similar | Similar |
| PMF | |||
| Clinical | Younger, lower WBC count, higher Hg/Hct, higher platelets | Younger, higher Hg/Hct, higher platelets | Younger, higher Hg/Hct, higher platelets |
| Thrombosis | Similar, possibly decreased risk | Similar | Similar |
| Leukemic transformation | Similar | Similar | Improved |
| Overall prognosis | Improved* | Similar | Improved |
| CALR vs JAK2 | CALR vs MPL | CALR vs triple negative | |
|---|---|---|---|
| ET | |||
| Clinical | Younger, male predominance, lower WBC count, lower Hg/Hct, higher platelets | Male predominance, otherwise similar | Male predominance, higher platelets |
| Thrombosis | Decreased risk | Decreased risk | Similar |
| Post-ET MF | Similar to increased | Similar | Similar |
| Overall prognosis | Similar | Similar | Similar |
| PMF | |||
| Clinical | Younger, lower WBC count, higher Hg/Hct, higher platelets | Younger, higher Hg/Hct, higher platelets | Younger, higher Hg/Hct, higher platelets |
| Thrombosis | Similar, possibly decreased risk | Similar | Similar |
| Leukemic transformation | Similar | Similar | Improved |
| Overall prognosis | Improved* | Similar | Improved |
Hct, hematocrit; Hg, hemoglobin; MF, myelofibrosis; WBC, white blood cell.
Improved overall survival may be restricted to type 1–like mutations.
Since the initial discovery of CALR mutations in MPN, there have been multiple studies investigating disease outcomes in terms of thrombosis, myelofibrotic/leukemic transformation, and overall survival (OS). There is robust evidence indicating improved thrombosis-free survival in CALR-mutant ET, with a twofold decreased risk for venous and arterial thrombosis compared with JAK2 V617F patients.7,33,34,40-43 CALR-mutant ET patients also appear to have improved thrombosis-free survival compared with MPL-mutant, but not triple-negative, ET patients.33,34 The data are less clear in PMF, although studies have also suggested a decreased risk for thrombosis.35,36 The risk of transformation to post-ET myelofibrosis (MF) seems to be similar between CALR-mutant and JAK2-mutant patients,7,33,34 although some studies have reported an increased risk for post-ET progression to MF in CALR-mutant patients.10,44 Similarly, the risk of blast transformation and leukemic progression in PMF patients appears to be mixed, with studies showing improved or similar leukemia-free survival in CALR-mutant patients.34-36,45 The disparate results in PMF are likely a reflection of overall smaller sample sizes and fewer events. In ET, OS is similar between CALR and JAK2, MPL, and triple-negative patients.7,8,33,40,41,43 However, unlike in ET, CALR mutation status has consistently emerged as an independent predictor of OS in PMF,8,34,35,40 which has also been borne out in meta-analyses.36 Indeed, median OS is estimated to be ∼17 years in CALR-mutant PMF patients compared with 9 years in JAK2-mutant PMF patients and 3 years in triple-negative PMF patients.35 This improved prognosis also applies to CALR PMF patients who subsequently receive hematopoietic stem cell transplantation,46,47 as well as in post-ET MF patients specifically.37
In addition, there are significant clinical and prognostic differences depending on the type of CALR mutation (Table 2). Type 1–like mutations are significantly more common in PMF, whereas type 2–like mutations are more common in ET.11,48 Phenotypic differences are also borne out in mice, because type 1–like engrafted mice display significantly more myelofibrosis than type 2–like engrafted mice.15,49 Within ET, type 2–like CALR patients have higher platelet counts; otherwise, both groups of patients display similar outcomes, including OS.48,50,51 The risk of MF progression may be higher in type 1 patients,11 consistent with its overall increased prevalence in PMF in general. Within PMF, type 1–like patients have significantly improved OS compared with type 2–like patients, with more similar clinical presentations and prognosis between type 2–like patients and JAK2-mutant patients.11,48,51-54 The improved prognosis of CALR in PMF may actually be restricted to type 1–like CALR mutations.52 Phenotypic differences in type 1–like vs type 2–like MPN patients may be related to the differential strength of MPL signaling activation and/or to the greater loss of calcium binding sites seen with type 1–like mutations.11
Summary of clinical features and outcomes of type 1–like vs type 2–like CALR mutations
| Type 1–like CALR | Type 2–like CALR | |
|---|---|---|
| Most common mutation | 52-bp deletion (L367fs*46) | 5-bp insertion (K385fs*47) |
| Prevalence | More common in MF | More common in ET |
| Clinical (ET) | Similar; lower platelet counts vs type 2 like | Similar; higher platelet counts vs type 1 like |
| Clinical (MF) | Less splenomegaly, leukocytosis, anemia, and circulating blasts; lower DIPSS score; higher platelets (all vs type 2 like) | More splenomegaly, leukocytosis, anemia, and circulating blasts; higher DIPSS score; lower platelets (all vs type 1 like). More similar to JAK2 V617F |
| Post-ET MF | Similar to/increased vs type 2 like | Similar to/decreased vs type 1 like |
| Overall prognosis (ET) | Similar to type 2 like | Similar to type 1 like |
| Overall prognosis (MF) | Improved vs type 2 like and JAK2 V617F | Worsened vs type 1 like; more similar to JAK2 V617F |
| Type 1–like CALR | Type 2–like CALR | |
|---|---|---|
| Most common mutation | 52-bp deletion (L367fs*46) | 5-bp insertion (K385fs*47) |
| Prevalence | More common in MF | More common in ET |
| Clinical (ET) | Similar; lower platelet counts vs type 2 like | Similar; higher platelet counts vs type 1 like |
| Clinical (MF) | Less splenomegaly, leukocytosis, anemia, and circulating blasts; lower DIPSS score; higher platelets (all vs type 2 like) | More splenomegaly, leukocytosis, anemia, and circulating blasts; higher DIPSS score; lower platelets (all vs type 1 like). More similar to JAK2 V617F |
| Post-ET MF | Similar to/increased vs type 2 like | Similar to/decreased vs type 1 like |
| Overall prognosis (ET) | Similar to type 2 like | Similar to type 1 like |
| Overall prognosis (MF) | Improved vs type 2 like and JAK2 V617F | Worsened vs type 1 like; more similar to JAK2 V617F |
Current treatment of CALR-mutant MPN patients
There are no rationally designed treatments targeted toward the CALR mutation. Standard cytoreductive therapies in MPNs, such as hydroxyurea, interferon-α, and ruxolitinib, have shown similar improvement in patients’ cell counts and symptoms, regardless of mutation status. Within PMF, a retrospective analysis of the COMFORT-II study confirmed no significant differences in response rates to ruxolitinib between mutant CALR-positive and CALR-negative patients.55 Similar to JAK2-mutant patients, treatment with ruxolitinib results in decreased spleen size and symptom palliation, but without a reduction in mutant CALR allele burden.55 CALR-mutant ET patients also demonstrate clinical and molecular responses to interferon-α therapy.56 One small retrospective study has suggested an inferior platelet response to anagrelide in CALR-mutant ET patients compared with JAK2-mutant ET patients; however, this finding needs to be replicated.57
CALR mutations impact clinical risk stratification and, thus, initial treatment decisions.58,59 Within PMF, absence of CALR type 1–like mutations confers higher-risk scores in MIPSS70 and GIPSS, which has implications for the timing of stem cell transplantation.38,39,60 The JAK2 V617F mutation confers higher thrombotic risk in the International Prognostic Score for Essential Thrombocythemia system.61 In certain young CALR-mutant ET patients, thrombotic risk may be sufficiently low such that these patients can be managed with observation alone, without the addition of aspirin. According to 1 retrospective study, aspirin use was not shown to decrease thrombosis in these patients and may even incur an increased risk for bleeding.62 Aspirin should generally be avoided in patients with acquired von Willebrand deficiency from extreme thrombocytosis, in which case cytoreductive therapies are considered to reduce bleeding risk.
Toward therapeutic targeting of mutant CALR
Insights into the mechanistic basis of mutant CALR-induced MPN reveal several potential novel therapeutic approaches (Figure 1). In particular, the mutant-specific C terminus of mutant CALR is attractive for immunological targeting. The presence of cell surface mutant CALR expression has highlighted the potential for a mutant-specific anti-CALR therapeutic antibody that could disrupt MPL activation.19,24 A second immunologic strategy is through targeting neoepitopes in the novel mutant CALR C terminus in the context of T-cell activation or engineered T-cell receptor–mediated immune therapy. This approach is complicated by the issue of HLA restriction and a current lack of evidence for natural major histocompatibility complex (MHC) processing and presentation of mutant CALR neoepitopes.63 CALR has a role in MHC class I (MHC-I) assembly; in experimental systems in which mutant CALR is overexpressed, impaired peptide loading in MHC-I antigen presentation occurs, resulting in downregulation and decreased stability of MHC-I on the cell surface.64 Consistent with this, stimulated CD8+ T-cell responses against mutant CALR epitopes are lacking in CALR-mutant patient samples.63,65 However, mutant CALR-specific CD4+ memory T-cell responses have been demonstrated in healthy individuals, suggesting that mutant CALR is immunogenic and immune escape occurs in patients with mutant CALR-driven MPN.66 A CD4+ T-cell clone with specific cytotoxicity against autologous CALR-mutant cells has been generated,65 and these results have formed the basis of a phase 1 vaccination study in Denmark with a CALR exon 9 peptide vaccine (NCT03566446). More recent evidence indicates that T cells from MPN patients express immune checkpoint molecules indicative of a T-cell exhaustion phenotype.67 Ex vivo treatment with an anti–PD-1 antibody rendered these T cells more responsive to mutant CALR peptide stimulation, suggesting that it may be possible to activate autologous T cells from CALR-mutant MPN patients to recognize and target mutant CALR neoepitopes.67 Additional immunological studies are required to determine whether mutant CALR neoepitopes are processed and presented by MHC, and clinical studies are needed to determine whether T cells can recognize and target these neoepitopes in vivo.
Other novel therapeutic approaches under consideration include a synthetic peptide to competitively inhibit mutant CALR-MPL binding, which has demonstrated some in vitro efficacy and synergy with JAK2 inhibitors.28 Further preclinical studies to optimize therapeutic delivery of such a peptide are necessary, especially because mutant CALR-MPL binding occurs inside the cell. Crystal structures of mutant CALR and the extracellular domain of MPL are also currently lacking, which hinders precise knowledge regarding their physical interaction.
Conclusions
In the almost 6 years since the identification of CALR mutations the field has progressed rapidly. We now have a more detailed understanding of how a mutated chaperone protein can result in MPN pathogenesis. Novel therapeutic approaches exploiting this mechanistic understanding are currently in development and will continue to expand as investigation into mutant CALR advances.
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL131835, the Starr Cancer Consortium (grant I9-A9-059), the MPN Research Foundation, and Gabrielle’s Angel Foundation for Cancer Research (all to A.M.). A.M. is a scholar of the Leukemia & Lymphoma Society. G.S.H. is supported by a K12 award from the National Institutes of Health, National Cancer Insititute (K12CA087723), a Sanchez and Ferguson Research Faculty Award, and an American Society of Hematology–Harold Amos Medical Faculty Development Program award.
Authorship
Contribution: A.M. conceived the manuscript idea and edited the manuscript; J.H. wrote the manuscript with input from G.S.H. and A.M.
Conflict-of-interest disclosure: A.M. has received honoraria from Blueprint Medicines, Roche, and Incyte for invited lectures, has served on an advisory board for CTI BioPharma, and receives research support from Janssen Pharmaceuticals. G.S.H. has served on advisory boards for Agios, Celgene, Incyte, and Jazz Pharmaceuticals and has received research support from Bayer, Incyte, and Merck. J.H. declares no competing financial interests.
Correspondence: Ann Mullally, Harvard Institutes of Medicine Building, Room 738, 77 Ave Louis Pasteur, Boston, MA 02115; e-mail: amullally@partners.org.
