

TO THE EDITOR:
In recent years, detection of circulating tumor plasma cells (CTPC), tumor cell–derived deoxyribonucleic acid (DNA), RNA, or protein markers in blood has gained interest for disease monitoring in multiple myeloma (MM).1,2 This is mainly because of (1) the minimally invasive nature of blood vs bone marrow (BM) analyses, (2) the possibility for more precise quantification of absolute numbers of CTPC than BM minimal residual disease (MRD) resulting from absence of potential hemodilution, and (3) the (nonlinear) correlation observed between CTPC numbers and BM disease burden at diagnosis.1,3 Recently, we have shown by high-sensitivity next-generation flow (NGF) that CTPC are systematically present in blood of MM at diagnosis, with an adverse prognostic impact for higher counts.3 These results highlight the relevance of greater levels of disease dissemination via blood in conferring a malignant behavior to MM, suggesting the presence of blood CTPC might be required for subsequent disease progression of treated MM patients.
Based on this hypothesis, here we investigate for the first time the prognostic impact of CTPC by NGF in blood of 137 newly diagnosed MM patients after active treatment outside clinical trials (supplemental Table 1 on the Blood Web site), in parallel to BM MRD and serum immunofixation (sIF). Overall, a total of 328 samples were analyzed: 274 paired BM and blood samples, plus 54 follow-up blood specimens. Following the EuroFlow-NGF MM MRD approach,4 a median (range) of 6 mL (3-14 mL) of blood and 1.8 mL (0.3-5 mL) of BM sample were lysed to (systematically) obtain ≥107 cells per sample. In parallel, sIF was measured by the HYDRAGEL kit (HYDRASYS system, Sebia, Barcelona, Spain).5 Statistical significance was set at P values < .05 (supplemental Materials). All studies were approved by the institutional review board.
Following therapy, persistence of CTPC in blood was detected in 26% of MM cases. This represents a 50% higher frequency than previously reported by conventional flow cytometry (18%-19%),6-8 reaching rates similar to those found with other high-sensitivity techniques such as allele-specific oligonucleotide polymerase chain reaction (25%-28.8%9,10 ) or next-generation sequencing (31%-34%2,11 for cell-free DNA and 40%2 for genomic leukocyte DNA). This translated into even higher differences among patients who reached complete response (CR)/stringent CR (sCR): 17% CTPC+ cases in our series vs 0%12,13 to <8%6,8 in other previous conventional flow cytometry studies (supplemental Table 2).
Despite the greater sensitivity and rate of positivity for CTPC reported here, a significant proportion of our MM cases that were BM MRD+ or sIF+ still had undetectable CTPC in (paired) blood samples: 55/137 (40%) and 41/137 (30%), respectively. In contrast, 15/36 (42%) CTPC+ cases were also sIF− (supplemental Table 2). These findings indicate that CTPC is a less sensitive MRD marker in MM than BM MRD, complementary to sIF, in line with previous observations.1 However, although BM MRD and sIF mainly reflect persistence of resistant tumor14 and tumor cell–derived immunoglobulins,15 they fail to provide insight on the ability of these cells to support tumor regrowth and/or dissemination, which ultimately determine disease progression. In contrast, CTPC might not only reflect tumor load but, particularly, the ability of persisting tumor cells to disseminate the disease and support tumor growth and progression at (multiple) distant sites in BM and other tissues, as previously suggested16 based on their more immature and prominent stem cell-like PC features compared with (paired) BM-derived tumor-plasma cells (TPC).3
Despite all of this, every CTPC+ case in our cohort was BM MRD+, suggesting that the presence of blood CTPC after therapy might be a surrogate marker of persistent BM MRD in guiding (eg, avoiding) subsequent (more invasive) BM aspiration procedures, particularly among sCR/CR patients. In contrast, a significant fraction of our CTPC− cases were BM MRD+ and/or sIF+, supporting the notion that MM is a BM disease with greater levels of infiltration by (usually) functional PC in BM vs PB. Prolonged half-life (∼23 days) and complete clearance (∼29 weeks) of the M-protein for the most prevalent immunoglobulin G subclass,17 in addition to persistence of extramedullary disease18 and/or the administration of monoclonal antibody-therapy (eg, daratumumab)19 for MM patients, might also explain sIF positivity in at least a subset of BM MRD−/sIF+ cases. Additionally, poor BM sample quality (eg, from hemodilution) might also play a role because abnormally low (≤0.002%)4 mast cell counts were detected here in 5/10 BM MRD−/sIF+ cases. In contrast, sIF negativity among 4 of our non-sCR/CR patients could be related to the appearance/persistence of plasmacytomas18 (2/4 cases), and high free light chain ratio levels (>500) without measurable M-component in serum and urine18 (1/4 cases), together with a non-secretory TPC15 detectable here in another MM patient.
From the prognostic point of view, our results based on real-world MM show for the first time that the absence vs presence of blood CTPC by NGF is a new powerful independent prognostic marker for progression-free survival (PFS) measured from the time of BM-MRD/CTPC assessment both among the entire MM patient cohort (hazard ratio [HR], 5.1; 95% confidence interval [CI], 2.9-8.9; P < .0001) (Figure 1A) and within sCR/CR cases (HR, 7.4; 95% CI, 3.0-18.2; P < .0001; Figure 1B), complementary to currently available prognostic tools such as sIF and BM MRD, respectively (Table 1), and regardless of the treatment phase BM-MRD/CTPC being assessed (supplemental Figure 1). Based on those covariables that showed a (statistically) significant effect on PFS in multivariate analysis (Table 1), a prognostic score was built that allowed identification of a subgroup of blood CTPC+ MM patients with a very poor outcome (score 2 in the risk stratification models proposed here) with PFS rates at 2 years of only 1% for the entire patient cohort (Figure 1G) and of 33% for sCR/CR MM patients (Figure 1H), respectively.
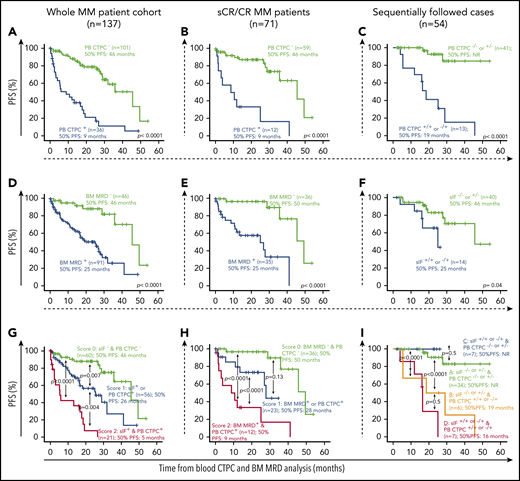
Prognostic impact of blood CTPC by NGF (vs BM MRD and sIF) on PFS of MM patients according to patient response to therapy. (A-B) Effect of PB CTPC, (D-E) BM MRD, (H) combination of both parameters, and (G) PB CTPC together with sIF status on PFS is displayed for (A, D, G) the entire MM cohort and (B, E, H) for sCR and CR patients, respectively. PFS curves of MM patients grouped according to (C) their sequential PB CTPC (−/− or +/− vs −/+ and +/+), (F) sIF status, or (I) a combination of both, are shown. Overall, CTPC− and MRD− was defined as the absence of TPC in PB or BM by NGF, respectively, with a limit of detection of <2 × 10−6. CR, complete response; NR, not reached; PB, peripheral blood.
Prognostic impact of blood CTPC by NGF (vs BM MRD and sIF) on PFS of MM patients according to patient response to therapy. (A-B) Effect of PB CTPC, (D-E) BM MRD, (H) combination of both parameters, and (G) PB CTPC together with sIF status on PFS is displayed for (A, D, G) the entire MM cohort and (B, E, H) for sCR and CR patients, respectively. PFS curves of MM patients grouped according to (C) their sequential PB CTPC (−/− or +/− vs −/+ and +/+), (F) sIF status, or (I) a combination of both, are shown. Overall, CTPC− and MRD− was defined as the absence of TPC in PB or BM by NGF, respectively, with a limit of detection of <2 × 10−6. CR, complete response; NR, not reached; PB, peripheral blood.
Multivariate analysis of prognostic factors For PFS In MM
| Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|
| Median PFS (mo) | P | HR | (95% CI) | P | |
| Prognostic factors for entire MM series | |||||
| Age | |||||
| <65 y | 28 | .3 | — | — | — |
| ≥65 y | 36 | ||||
| Cytogenetic profile by FISH | |||||
| Standard-risk | 36 | .07 | — | — | — |
| High-risk | 16 | ||||
| Serum IF | |||||
| Negative | 41 | .001 | — | — | — |
| Positive | 18 | 2.4 | (1.3-4.4) | .004 | |
| BM MRD status by NGF | |||||
| Negative | 46 | <.0001 | — | — | — |
| Positive | 25 | ||||
| PB CTPC status by NGF | |||||
| Negative | 46 | <.0001 | — | — | — |
| Positive | 9 | 5.1 | (2.9-8.9) | <.0001 | |
| Prognostic factors for sCR/CR cases | |||||
| Age | |||||
| <65 y | 50 | .5 | — | — | — |
| ≥65 y | 41 | ||||
| Cytogenetic profile by FISH | |||||
| Standard-risk | 50 | .09 | — | — | — |
| High-risk | 28 | ||||
| BM MRD status by NGF | |||||
| Negative | 50 | <.0001 | — | — | — |
| Positive | 25 | 6.1 | (1.5-24.4) | .01 | |
| PB CTPC status by NGF | |||||
| Negative | 46 | <.0001 | — | — | — |
| Positive | 9 | 7.4 | (3.0-18.2) | <.0001 | |
| Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|
| Median PFS (mo) | P | HR | (95% CI) | P | |
| Prognostic factors for entire MM series | |||||
| Age | |||||
| <65 y | 28 | .3 | — | — | — |
| ≥65 y | 36 | ||||
| Cytogenetic profile by FISH | |||||
| Standard-risk | 36 | .07 | — | — | — |
| High-risk | 16 | ||||
| Serum IF | |||||
| Negative | 41 | .001 | — | — | — |
| Positive | 18 | 2.4 | (1.3-4.4) | .004 | |
| BM MRD status by NGF | |||||
| Negative | 46 | <.0001 | — | — | — |
| Positive | 25 | ||||
| PB CTPC status by NGF | |||||
| Negative | 46 | <.0001 | — | — | — |
| Positive | 9 | 5.1 | (2.9-8.9) | <.0001 | |
| Prognostic factors for sCR/CR cases | |||||
| Age | |||||
| <65 y | 50 | .5 | — | — | — |
| ≥65 y | 41 | ||||
| Cytogenetic profile by FISH | |||||
| Standard-risk | 50 | .09 | — | — | — |
| High-risk | 28 | ||||
| BM MRD status by NGF | |||||
| Negative | 50 | <.0001 | — | — | — |
| Positive | 25 | 6.1 | (1.5-24.4) | .01 | |
| PB CTPC status by NGF | |||||
| Negative | 46 | <.0001 | — | — | — |
| Positive | 9 | 7.4 | (3.0-18.2) | <.0001 | |
High-risk cytogenetics was defined as presence at diagnosis of t(4;14); t(11;14); t(14;16); 1q amplification, deletion 13q, and/or deletion 17p. Standard-risk cytogenetics includes all other cases. (Data available in 96/137 cases.)
FISH, fluorescent in situ hybridization; IF, immunofixation.
These results, together with the demonstration that CTPC are systematically detected in blood of MM at diagnosis3 and at relapse,13 suggest that detection of blood CTPC rather than a surrogate marker of response is a strongly reliable predictor of impending (early) disease progression9 (Figure 1A-B). In contrast, BM MRD would be a better predictor of persistent disease (Figures 1D-E) and longer term prognosis of MM undergoing different therapies, as recurrently shown in the literature in the settings of clinical trials, both for conventional20-22 and for high-sensitivity NGF4,23 and next-generation sequencing24 approaches. However, frequent BM sampling is hampered by the invasive nature of BM aspiration procedures,1 whereas more frequent follow-up of CTPC in blood is feasible. Thus, sequential monitoring of blood CTPC was performed in a subset of 54 cases in parallel to sIF. Our results showed that MM patients who were persistently CTPC− (CTPC−/−) or that became CTPC− after being CTPC+ (CTPC+/−) showed a significantly better outcome than cases with a blood CTPC+ result in the last follow-up study (CTPC+/+ or −/+) (Figure 1C), independent of sIF status (Figure 1F, I). However, this should be confirmed in larger series of patients with longer monitoring because the limited number of sIF+/+ or −/+ (n = 7) cases was observed for a paradoxically (slightly) better outcome vs the sIF−/− or +/− (n = 34) MM showing no CTPC in the last follow-up study (ie, CTPC−/− or +/− cases) (Figure 1I). In spite of that, the former findings suggest that blood CTCP might provide additional relevant prognostic information to single time-point BM MRD assessment in predicting for longer term outcome in patients that either (persistently) remain or turn CTPC− in sequential follow-up studies, in line with previous reports.9,25
In summary, we show here that blood CTPC is a novel independent prognostic marker for PFS in real-world MM prone to more frequent monitoring, which provides early indication of impending disease progression, regardless of BM MRD and sIF status. These results suggest that presence of blood CTPC of MM after therapy probably reflects a unique distinct tumor biology (eg, tumor dissemination capacity) at a given time point after therapy with important clinical consequences. Further studies in larger series of MM patients outside and inside clinical trials are required to confirm our findings.
For original data, please contact orfao@usal.es.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Sergio Matarraz and Lourdes Martín-Martín for their support on statistical analysis.
This work has been supported by the International Myeloma Foundation-Black Swan Research Initiative and the EuroFlow Consortium; Centro de Investigación Biomédica en Red de Cáncer (CIBER-ONC; Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, Madrid, Spain, and FONDOS Europeo de Desarrollo Regional) grants CB16/12/00400 (L.S.-F., J.F.-M., A.C.-M., and A.O.), CB16/12/00233 (N.P., O.G.-S., M.D.-C., M.G. and M.-V.M.), CB16/12/00369 and CB16/12/00489 (B.P. and J.S.M.); grant SA079U14 from the Consejería de Educación, Junta de Castilla y León, Valladolid, Spain; and grant DTS15/00119 from the Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, Madrid, Spain; by the Research Foundation of the State of Rio de Janeiro, Rio de Janeiro, Brazil (grant E26/110.105/2014) and Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPQ of Brazil (grants 303765/2018-6 and 409440/2016-7).
The EuroFlow Consortium is an independent scientific consortium that aims at innovation and standardization of diagnostic flow cytometry. All consortium members that contributed are listed as authors. All acquired knowledge and experience within EuroFlow is shared with the scientific and diagnostic community after protection of the relevant Intellectual Property; for example, by filling patents. The involved patents are owned by the EuroFlow Consortium and licensed to companies, including Cytognos SL (Salamanca, Spain), Becton/Dickinson Biosciences (San José, CA), and Immunostep SL (Salamanca, Spain). The revenues of the patents are exclusively used for EuroFlow Consortium activities, such as for covering (in part) the costs of the Consortium meetings, the EuroFlow Educational Workshops, and the purchase of custom-made reagents for collective experiments. J.F.-M., J.J.M.v.D., and A.O. are part of the inventors on the EuroFlow-owned patent PCT/NL/2013/050420; US 62/072 498 (Methods, reagents and kits for detecting minimal residual disease). This patent is licensed to Cytognos, which pays royalties to the EuroFlow Consortium.
Authorship
Contribution: L.S.-F., T.C.S., R.P., A.C.-M., and O.G.S. performed experiments; L.S.-F. and J.F.M. analyzed results; N.P., M.D.-C., R.J.P.d.M., L.G.-M., J.M.A.-A., A.G.-M., C.A.-F., J.L., A.B.-G., A.M., E.S.d.C. and M.G. contributed to the data collection and performed patients management; L.S.-F., J.F.-M., B.P., J.S.M., M.V.M., B.D., J.J.M.v.D., and A.O. designed the research; and L.S.-F., J.F.-M. and A.O. wrote the paper.
Conflict-of-interest disclosure: M.-V.M. declares honoraria for lectures and advisory boards from Janssen, Celgene, Amgen, Takeda, Abbvie, GSK, Adaptive, EDO-Mundipharma, and Pharmamar. The remaining authors declare no competing financial interests.
Correspondence: Alberto Orfao, Centro de Investigación del Cáncer (CSIC-USAL), Avenida Universidad de Coimbra S/N, Campus Miguel de Unamuno, Salamanca 37007, Spain; e-mail: orfao@usal.es.
Appendix: study group members
The members of the EuroFlow Consortium are: J.J.M.v.D., W. M. Bitter, B. R. Lubbers, A. S. M. van der Meij, C. I. Teodosio, M. Zlei, A. J. van der Sluijs-Gelling, M. van der Burg (Leiden University Medical Center); V. H. J. van der Velden, A. W. Langerak, J. te Marvelde, J. Schilperoord-Vermeulen, A. Blijkerk, K. C. Heezen (Erasmus MC); A.O., J. Almeida, M. B. Vidriales, J.F.-M., M. Pérez-Andrés, S. Matarraz, E. Blanco, L. Martín, Q. Lecrevisse, J. J. Pérez-Morán, N. Puig (University of Salamanca); A. Medina Almeida, M. Gomes da Silva, T. Faria (Instituto Portugués de Oncologia); M. Brüggemann, M. Ritgen, M. Szczepanowski, S. Kohlscheen, A. Steinert, E. Harbst, J. Finke (University of Schleswig-Holstein); V. Asnafi, L. Lhermitte, E. Duroyon (Hôpital Necker-Enfants Malades); J. Trka, O. Hrusak, T. Kalina, E. Mejstrikova, M. Novakova, D. Thurner, V. Kanderova (Charles University); T. Szczepanski, L. Sędek, J. Bulsa, L. Slota, J. Kulis (Medical University of Silesia); C. E. Pedreira, E. Sobral da Costa (Federal University of Rio de Janeiro); S. Nierkens, A. de Jong, A. de Koning (Dutch Childhood Oncology Group); M. Lima, A. H. Santos (Centro Hospitalar do Porto/University of Porto); S. Böttcher, S. Lange, R. Engelmann, D. Paape, C. Machka (Universitätmedizin Rostock); G. Gaipa, C. Burracchi, C. Bugarin (Università di Milano); E. Lopez-Granados, L. del Pino Molina (University Hospital La Paz-IdiPAZ); M. Vlkova, J. Nechvatalova (St Anne's Faculty Hospital); M. Roussel (University of Rennes); L. Campos-Guyotat, C. Aanei (CHU de Saint-Etienne); J.S.M., B.P., L. Burgos (Universidad de Navarra); N. Villamor-Casas, L. Magnano (Hospital Clínic de Barcelona); J. Philippé, C. Bonroy, B. Denys, A. Willems, P. Breughe, J. de Wolf (University Hospital Ghent); A. E. Sousa, S. L. Silva (University of Lisbon); P. Fernandez, D. Morf (Kantonsspital Aarau).

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal