


Abstract
Allogeneic stem cell transplantation is a cornerstone of curative therapy for high-risk and/or advanced hematological malignancies but remains limited by graft-versus-host disease (GVHD). GVHD is initiated by the interaction between recipient antigen-presenting cells (APCs) and donor T cells, culminating in T-cell differentiation along pathogenic type-1 and type-17 paradigms at the expense of tolerogenic regulatory T-cell patterns. Type-1 and type-17 T cells secrete cytokines (eg, granulocyte-macrophage colony-stimulating factor and interferon-γ) critical to the cytokine storm that amplifies expansion of donor APCs and their alloantigen presentation. It has become increasingly clear that pathogenic donor T-cell differentiation is initiated by both professional recipient APCs (eg, dendritic cells [DCs]) and nonprofessional APCs (eg, epithelial and mesenchymal cells), particularly within the gastrointestinal (GI) tract. In the immediate peritransplantation period, these APCs are profoundly modified by pathogen-associated molecular pattern (PAMP)/damage-associated molecular pattern (DAMP) signals derived from conditioning and intestinal microbiota. Subsequently, donor DCs in the GI tract are activated by DAMP/PAMP signals in the colon that gain access to the lamina propria once the mucosal barrier mucosa is compromised by GVHD. This results in donor DC expansion and alloantigen presentation in the colon and subsequent migration into the mesenteric lymph nodes. Here, new donor T cells are primed, expanded, differentiated, and imprinted with gut-homing integrins permissive of migration into the damaged GI tract, resulting in the lethal feed-forward cascade of GVHD. These new insights into our understanding of the cellular and molecular factors initiating GVHD, both spatially and temporally, give rise to a number of logical therapeutic targets, focusing on the inhibition of APC function in the GI tract.
Introduction
Allogeneic hematopoietic stem cell transplantation (alloSCT) is an established curative therapy for both nonmalignant (eg, immune deficiencies, errors of metabolism) and malignant hematological disorders. The curative potential of alloSCT for malignancy lies in the ability of donor T cells and natural killer cells to mount graft-versus-leukemia (GVL) responses that target multiple donor-host disparate alloantigens, hematopoietic antigens, or malignancy-specific antigens. In the last 2 decades, a number of small-molecule inhibitors have become available that target malignant driver kinase or dehydrogenase mutations (eg, flt3, bcr-abl, idh-2). Most recently, gene-modified chimeric antigen receptor T-cell therapies have emerged, targeting, in particular, CD19+ B-cell malignancies. Although these new agents offer remarkable disease specificity, their place and timing in the treatment of malignant disease continue to be explored, including in combination with alloSCT. Furthermore, because these agents usually target single pathways, immune escape is not uncommon, meaning that GVL effects after alloSCT that target multiple antigens remain an important cornerstone of curative therapy.
The major limitations of alloSCT remain graft-versus-host disease (GVHD) and disease relapse, the latter a process whereby GVL fails. GVHD is the process whereby recipient, and later donor, antigen-presenting cells (APCs) present recipient alloantigen to naïve donor T cells to generate an immune response characterized by target tissue damage.1 Importantly, recipient hematopoietic APCs are short lived as a result of their eradication by chemoradiotherapy during conditioning and the GVHD response.2 In contrast, recipient nonhematopoietic APCs are present in perpetuity (Figure 1). Because the T cells responsible for GVHD and GVL are often indistinguishable, the separation of these 2 detrimental vs beneficial immune responses is difficult. GVHD and GVL are absent after autologous and syngeneic SCTs,3 where no donor-recipient alloantigen disparity exists, as well as after stringently T-cell–depleted alloSCT, where mature donor T cells capable of mediating a response to alloantigen are absent.4 Therefore, understanding the mechanisms by which naive T cells recognize and respond to alloantigen on various APCs is critical to modulating GVHD and GVL responses for therapeutic gain.
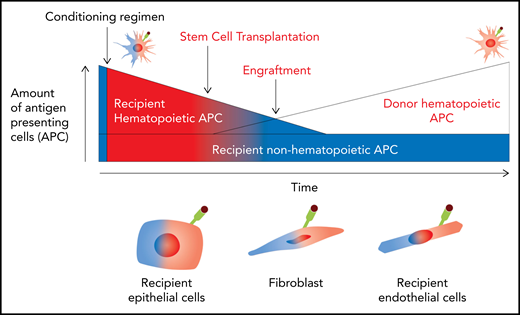
Pathways of antigen presentation during alloSCT. Before conditioning, all APCs are recipient and noninflamed (blue). After conditioning, damage-associated molecular pattern (DAMP)/pathogen-associated molecular pattern (PAMP) signals promote antigen presentation by recipient hematopoietic and nonhematopoietic APCs (red). After transplantation, recipient hematopoietic APCs are rapidly eradicated by conditioning and GVHD and donor APC constitute. In contrast, nonhematopoietic APCs continue in the host indefinitely, although their antigen presentation capacity is dependent on environmental cues, particularly the presence of active inflammation.
Pathways of antigen presentation during alloSCT. Before conditioning, all APCs are recipient and noninflamed (blue). After conditioning, damage-associated molecular pattern (DAMP)/pathogen-associated molecular pattern (PAMP) signals promote antigen presentation by recipient hematopoietic and nonhematopoietic APCs (red). After transplantation, recipient hematopoietic APCs are rapidly eradicated by conditioning and GVHD and donor APC constitute. In contrast, nonhematopoietic APCs continue in the host indefinitely, although their antigen presentation capacity is dependent on environmental cues, particularly the presence of active inflammation.
Key to the potential dissociation of GVL from GVHD are the differential spatial and temporal requirements for the 2 processes. Acute GVHD is characterized by apoptosis in the skin, liver, and gastrointestinal (GI) tract relatively early (within 6 months) after alloSCT, whereas chronic GVHD (cGVHD) is characterized by fibrosis and occurs later after alloSCT (beyond 3 months), typically in the skin, lung, and eyes/mouth.5,6 In contrast, a successful GVL effect must be present at the site where the hematological malignancy resides, typically the bone marrow (BM) and lymphoid organs, and is likely to be required long term. Conceptually, both GVHD and GVL are initiated by interactions between cognate peptide–major histocompatibility complex (MHC) and T-cell receptors (TCRs) on APCs and T cells, respectively, to drive T-cell differentiation, systemic inflammation, and memory T-cell differentiation.7,8 Although GVHD may be mediated by T-cell cytolytic pathways and soluble inflammatory (ie, cytokine) signals that do not require cognate interactions between T cells and target tissue MHC,9 GVL is strictly dependent on these cognate interactions.10 It is also important to remember that the initiation and effector phases of both GVHD and GVL are dependent on antigen presentation within MHC classes I (MHC-I) and II (MHC-II), which are not clearly distinct in time or space. With this in mind, we discuss recent advances in our understanding of the processes by which APCs initiate and regulate GVHD, with particular relevance to pathways that seem therapeutically tractable.
Recipient vs donor APCs: initiating, amplifying, and regulating acute GVHD
GVHD is the result of allogeneic disparity, either at the level of the MHC or minor histocompatibility antigen (mHA), which represent polymorphic proteins that differ in donor and recipient. mHAs are critical to the induction of GVHD when donor and recipient are MHC matched, but MHC disparity allows endogenous self-antigen (rather than mHA) to be presented in a conformation within that is recognized as an alloantigen (ie, molecular mimicry; reviewed by Koyama and Hill11 ) by T cells from MHC-disparate donors.12 Host alloantigen is presented by MHC-I or MHC-II to donor CD8+ T cells or donor CD4+ T cells, respectively, and antigen recognition by both of T-cell subsets is involved in the induction of acute GVHD, because depletion of either attenuates clinical GVHD.13-18 In contrast, pan–T-cell depletion preventSs GVHD, but at the expense of increased risk of graft rejection and relapse.19
To explore the mechanisms of GVHD, preclinical murine transplantation models have been developed that are CD8+ T cell mediated (MHC-I dependent) and/or CD4+ T cell mediated (MHC-II dependent). MHC-I is expressed on almost all nucleated cells, whereas MHC-II expression is restricted. Despite the ubiquitous expression of MHC-I, the cells that present alloantigen on MHC-I to initiate CD8-dependent GVHD are primarily host (recipient) hematopoietic (rather than nonhematopoietic) cells.1,20 Although 1 study20 suggested MHC-I–dependent GVHD could also be initiated by radioresistant (putatively nonhematopoietic) APCs, findings were consistent with the initial pivotal study, where the effects of hematopoietic APCs dominated.1
In contrast to MHC-I–dependent GVHD, the APCs that initiate MHC-II–dependent GVHD remain less clear. Host hematopoietic APCs are sufficient to induce MHC-II–dependent GVHD.9,21 This notion that host hematopoietic APCs are critical for the initiation of GVHD is further supported by studies in which MHC-I– or MHC-II–sufficient conventional dendritic cells (DCs) and DC subsets were adoptively transferred to MHC-deficient recipients at the time of alloSCT.22,23 In contrast, host B cells24 and host macrophages25,26 do not seem to initiate GVHD and instead regulate disease. However, the ability of nonhematopoietic APCs to induce CD4+ T-cell–mediated GVHD has not been addressed in depth because of the technical difficulty of using MHC-II–deficient BM chimeras that develop the lethal autoimmune disease as a result of a failure of thymic negative selection.27 In addition, durable selective depletion of host DCs was not possible in the initial transgenic mice (CD11c-DTR) expressing diphtheria toxin receptor under the control of CD11c promoter, because they did not tolerate repeated diphtheria toxin dosing (required for sustained DC and CD11c-expressing macrophage depletion).28,29 Several newer studies have suggested the ability of nonhematopoietic APCs to initiate GVHD in isolation. Li et al30 demonstrated that the profound depletion of host professional APCs, including conventional DCs, plasmacytoid DCs, tissue macrophages, and B cells, did not decrease CD4-dependent GVHD. A concurrent study used TCR transgenic donor T cells (TEa) and BM chimeras (H-2b/d) in which the specific MHC-II locus required to stimulate TEa cells (I-Ab) was deleted in hematopoietic cells while preserving I-A/I-Ed and thus thymic selection. These studies demonstrated that nonhematopoietic APCs alone could potently induce lethal CD4+ T-cell–mediated GVHD.31 Furthermore, host DCs seemed to attenuate CD4-dependent GVHD by inducing activation-induced cell death (AICD) of donor T cells.31 Consistent with this, the absence of MHC-II in host hematopoietic cells did not protect recipients from CD4+ T-cell–mediated GVHD.20 Subsequent studies of host DC depletion by other investigators have confirmed these results.32,33 The depletion of host DCs thus dramatically enhances mortality in CD4+ T-cell–mediated,31 unseparated T-cell–mediated,32 and, to a lesser extent, CD8+ T-cell–mediated GVHD.30 In mice, recipient CD8+ DCs seem to be the primary subset for this deletional process.32,33 Recipient macrophages mediate SIRPα-CD47 interaction–dependent phagocytosis of donor T cells to regulate GVHD,26 whereas recipient B cells do so via interleukin-10 (IL-10) secretion.24 However, many issues remain unresolved, including the mechanisms by which recipient DCs kill donor T cells and the pathways by which macrophages and B cells themselves recognize alloantigen. Nevertheless, this series of findings indicates the important role that nonhematopoietic APCs likely play in GVHD. However, without direct evidence as to which nonhematopoietic cell types initiate GVHD, it remained arguable that very low residual levels of intact MHC-sufficient host hematopoietic APCs in BM chimeric recipients may be sufficient to induce GVHD (as opposed to nonhematopoietic cells acting as APCs).
Nonhematopoietic APCs and GVHD
It has therefore been important to determine whether nonhematopoietic APC subsets can initiate MHC-II–dependent GVHD in their own right and, if so, to identify the factors controlling their acquisition of APC function. Although human and rodent epithelial cells, endothelial cells, and fibroblasts have been known to express MHC-II during inflammation,34-37 a direct pathogenic role for such cells has only recently been definitively shown.31,38 Indeed, this phenomenon has been considered a response to, rather than an important cause of, disease.39-42 In the lung, MHC-II antigen presentation by alveolar epithelial cells43 or nonhematopoietic cells44 has also been suggested to induce CD4+ regulatory T (Treg) cells and tolerance. It is important to note that antigen presentation in alloSCT is a completely different scenario to that during infection, because inflammation is reliably invoked during conditioning chemoradiotherapy, and alloantigen is ubiquitous and present indefinitely, including at environmental interfaces, where danger signals can potently augment immunity. Indeed, MHC-II expression on intestinal epithelial cells (IECs) has been demonstrated as tolerogenic in infectious colitis models45 and capable of modulating intestinal stem cell differentiation.46 It has recently been demonstrated that intestinal commensal microbiota induce MHC-II expression on IECs in the ileum at steady state and that this process is amplified immediately after conditioning.38 Furthermore, the presence of MHC-II on host IECs is sufficient to initiate CD4+ T-cell–mediated GVHD, whereas mesenchymal and endothelial cells have limited or no role, respectively.38 Importantly, the intestinal stem cell in the ileum, known to express MHC-I and MHC-II, has also been shown to be an early target of GVHD after transplantation, with CD4 T-cell infiltration seen in the crypt base within 3 days, even by intravital microscopy.47 The question of how rapid naïve T-cell migration into tissues occurs after alloSCT is important, because donor naïve T cells need to make cognate interaction with nonhematopoietic APCs in the GI tract to be primed at this site. Naïve T cells express CD62L and CCR7. The ligand of CD62L is MAdCAM-1, which is expressed on endothelial cells and in the inflamed bowel.48 Although CCL19 and CCL21, the ligands of CCR7, are known to be highly expressed in secondary lymphoid organs, high CCL19 expression in the inflamed colon (>40-fold of steady state) has been demonstrated in murine colitis models.49 We have demonstrated that naïve alloantigen-specific T cells become activated (within 24 hours) and attain an effector memory (CD44+CD62L−) phenotype (within 5 days) in lethally irradiated recipients, independent of alloantigen. Indeed, donor T cells can be found in the GI tract within 6 hours of transplantation.31 Thus, transplantation results in a unique scenario where T cells can rapidly access tissue sites as well as lymphoid tissue.
Together, these findings are important for a number of reasons: the targeting of therapy to host professional/hematopoietic APCs (ie, DCs) is insufficient to prevent GVHD and may make it worse; targeting antigen presentation by IECs may specifically diminish GVHD in the GI tract, while maintaining GVL effects, given that leukemic cells reside in the BM; and finally, the findings provide a link between the microbiome and the initiation of GVHD in the GI tract.
In contrast to recipient APCs, donor APCs in isolation are very inefficient at inducing acute GVHD.31,50 However, once GVHD is initiated by host APCs, donor APCs are potent rheostats of disease severity. In this process, tissue damage in the GI tract compromises the barrier integrity, allowing microbiome-derived PAMP signals and DAMP signals (eg, from dying cells) to activate donor-derived DCs in the colon, which promotes their expansion and capacity to present alloantigen at this site. These CD103+ DCs migrate to the mesenteric nodes under the guidance of CCR7, where they amplify donor T-cell proliferation and differentiation, while imprinting gut-homing receptor α4β7.51 These imprinted and differentiated cells thus immigrate into the GI tract to mediate lethal GVHD (Figure 2). Recently, it has been noted that pathogenic T cells secrete large amounts of granulocyte-macrophage colony-stimulating factor (GM-CSF), a cytokine that expands myeloid cells, including donor DCs.52,53 GM-CSF–secreting donor T cells of both T helper (Th)17 and non-Th17 lineages accumulate within the colon, and GM-CSF itself enhances donor DC expansion and alloantigen presentation in the mesenteric lymph nodes.54 Thus, host APCs initiate a T-cell differentiation paradigm that further feeds forward on this axis to directly expand GM-CSF–dependent donor DCs, which in turn ensure severe acute GVHD in the lower GI tract.51,54
![Alloantigen presentation in acute GVHD. After conditioning, antigen presentation is augmented in recipient hematopoietic APCs (eg, DCs) and nonhematopoietic APCs (eg, IECs in the ileum) via microbiota-driven cytokine signals (IL-12 from macrophages and interferon-γ [IFN-γ] from type-1 innate lymphoid cells [ILC1] and T cells). After transplantation, naïve donor T cells encounter alloantigen in the lymphoid organs (primarily presented by hematopoietic APCs) and in the GI tract (presented by hematopoietic APCs and nonhematopoietic APCs, including IECs) and undergo pathogenic (Th1/Th17) differentiation to initiate target tissue damage (GVHD). After engraftment, and in the presence of disrupted barrier function in the colon mediated by GVHD, donor colonic DCs expand and present large amounts of locally acquired alloantigen. These donor DCs migrate from the colon to the mesenteric lymph node under the influence of CCR7 and present alloantigen to new donor T cells, which undergo activation and Th1/Th17 differentiation under the influence of IL-12/IL-6. Finally, donor T cells are imprinted with α4β7 integrins, which guide their migration to the GI tract to perpetuate severe acute GVHD in a feed-forward cascade. Adapted from Koyama and Hill11 and Koyama et al38 with permission.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/24/10.1182_blood.2019000823/4/m_bloodbld2019000823cf2.png?Expires=1769117758&Signature=s2kr69Wh9XfNEAwqloB8Jdx3A9mAqZXx8Ski0Et0M76zn2UYpLIhg4xfSVPDXDIF-vWORJvjjOaSOsMwLCamF-8J1VgTpg0tU8teaRwBDMTxfdKSW9~PIpemGwHFPeWOt0ngUgIa4eMeOx8-PCoZbkQZx-RKfgmL7wpGirldcmSxXRpgg2jI1Y-ZCCcAcIR7zWoZxVhzHrsGiDUxoj4mGNkB-GrQD74bBWHI9FRABYb2vV1yX~f-NusRpB3YKm-Vz7vEMn4YwV3rmt8MH~IWJzTXOEi~-JQ2LqBdYvyqZQpfYA2~qix58CVP5TO0VB~5V1w-Wm9VrR-~HPenzTA3hA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Alloantigen presentation in acute GVHD. After conditioning, antigen presentation is augmented in recipient hematopoietic APCs (eg, DCs) and nonhematopoietic APCs (eg, IECs in the ileum) via microbiota-driven cytokine signals (IL-12 from macrophages and interferon-γ [IFN-γ] from type-1 innate lymphoid cells [ILC1] and T cells). After transplantation, naïve donor T cells encounter alloantigen in the lymphoid organs (primarily presented by hematopoietic APCs) and in the GI tract (presented by hematopoietic APCs and nonhematopoietic APCs, including IECs) and undergo pathogenic (Th1/Th17) differentiation to initiate target tissue damage (GVHD). After engraftment, and in the presence of disrupted barrier function in the colon mediated by GVHD, donor colonic DCs expand and present large amounts of locally acquired alloantigen. These donor DCs migrate from the colon to the mesenteric lymph node under the influence of CCR7 and present alloantigen to new donor T cells, which undergo activation and Th1/Th17 differentiation under the influence of IL-12/IL-6. Finally, donor T cells are imprinted with α4β7 integrins, which guide their migration to the GI tract to perpetuate severe acute GVHD in a feed-forward cascade. Adapted from Koyama and Hill11 and Koyama et al38 with permission.
Alloantigen presentation in acute GVHD. After conditioning, antigen presentation is augmented in recipient hematopoietic APCs (eg, DCs) and nonhematopoietic APCs (eg, IECs in the ileum) via microbiota-driven cytokine signals (IL-12 from macrophages and interferon-γ [IFN-γ] from type-1 innate lymphoid cells [ILC1] and T cells). After transplantation, naïve donor T cells encounter alloantigen in the lymphoid organs (primarily presented by hematopoietic APCs) and in the GI tract (presented by hematopoietic APCs and nonhematopoietic APCs, including IECs) and undergo pathogenic (Th1/Th17) differentiation to initiate target tissue damage (GVHD). After engraftment, and in the presence of disrupted barrier function in the colon mediated by GVHD, donor colonic DCs expand and present large amounts of locally acquired alloantigen. These donor DCs migrate from the colon to the mesenteric lymph node under the influence of CCR7 and present alloantigen to new donor T cells, which undergo activation and Th1/Th17 differentiation under the influence of IL-12/IL-6. Finally, donor T cells are imprinted with α4β7 integrins, which guide their migration to the GI tract to perpetuate severe acute GVHD in a feed-forward cascade. Adapted from Koyama and Hill11 and Koyama et al38 with permission.
Presentation of endogenous vs exogenous alloantigen
In terms of recipient APCs, alloantigen can be endogenously derived from intracellular (cytosolic) or exogenous (endocytosed/phagocytosed) sources. Although MHC-I primarily loads endogenous (intracellular) antigens and MHC-II loads exogenous (extracellular) antigens, MHC-I can present extracellular antigens (ie, cross presentation), and vice versa, MHC-II can present endogenous antigens (via the autophagy pathway).55 Alloantigen presentation by donor APCs requires the uptake, processing, and presentation of recipient antigens. This process is characteristic of MHC-II presentation by migratory DCs in the GI tract.51 In CD8-dependent, MHC-matched models of GVHD, there is an apparent requirement for endogenous presentation of mHAs by hematopoietic APCs.1 A subsequent study demonstrated that although host hematopoietic APCs primarily present endogenous/intracellular mHAs, this process may be modified by the availability of nonhematopoietic-derived mHAs.56 In contrast, lethal CD4-dependent GVHD ensues when host nonhematopoietic cells express mHAs, regardless of expression by hematopoietic APCs.57 This leads to 2 possible scenarios: host hematopoietic APCs acquire and present exogenous nonhematopoietic mHAs, or host nonhematopoietic APCs present primarily endogenous mHAs within MHC-II. Of note, we found transcriptional signatures in pathways of endoplasmic reticulum phagosomes and cross presentation of exogenous antigens in inflamed IECs,38 and the capacity for proteolytic degradation of exogenous antigen in IECs has also been demonstrated.46 Therefore, the potential contribution of exogenous antigen in nonhematopoietic APCs, including IECs, is very likely.
Effects of the microbiome on alloantigen presentation
A consistent finding in clinical and experimental alloSCTs is that myeloablative conditioning (MAC) is a significant risk factor in the timing and severity of acute GVHD relative to reduced-intensity conditioning (RIC).58-60 Of note, MAC induces rapid and complete donor chimerism, including reconstitution of hematopoietic APCs. If host hematopoietic APCs are essential for active GVHD, RIC, which eliminates host hematopoietic cells more slowly, might be predicted to result in protracted alloantigen presentation and enhanced GVHD. Indeed, in experimental systems, the induction of MHC-I–dependent GVHD requires host APCs to present alloantigen to donor CD8 T cells within a 5-day window of total-body irradiation to induce GVHD.2 Thus, although RIC may prolong the survival of host hematopoietic APCs after BM transplantation (BMT), it is balanced by lower levels of inflammation and APC activation than occur after MAC. Intensive conditioning promotes PAMP translocation into the systemic circulation, which increases tumor necrosis factor–dependent cytotoxicity by macrophages and enhances the capacity of DCs to drive Th1 differentiation.61-63 The crucial involvement of DAMP/PAMP signals in this process has been demonstrated in many studies: administration of LPS antagonists reduced GVHD64 ; cytosine-phosphorothioate-guanine oligodeoxynucleotides (mimicking viral and bacterial DNA), which ligate toll-like receptor 9 (TLR9), accelerated GVHD in an IFN-γ–dependent manner65 ; inhibition of IL-1 by anti–IL-1 receptor antibody or uric acid by uricase, which are involved in the inflammasome pathway, and deficiency of recipient inflammasome machinery (NLRP3 or ASC) decreased GVHD60,66 ; and interaction of P2X7R with soluble ATP enhanced GVHD.67 A study by Li et al68 was therefore striking, in that the intensive depletion of TLRs (MyD88 and TRIF), the inflammasome pathway (via MyD88), and type 1 IFN signals from host hematopoietic cells did not reduce GVHD. These studies did not exclude a role for DAMP/PAMP signals in the induction of GVHD via host nonhematopoietic cells or donor cells. Indeed, direct MyD88 signaling in donor T cells leads to Th1/Th17 or Tc1 differentiation and enhances GVHD in an ST2-dependent manner.69,70 Regarding the effects of DAMP/PAMP signals on APCs, it is likely that these signals are transmitted to both hematopoietic and nonhematopoietic APCs. Most recently, we have described the mechanism by which intestinal microbiota are responsible for MHC-II expression and antigen presentation by host IECs in the ileum.38 The absence of intestinal microbiota or MyD88/TRIF molecules in recipient tissue abrogated MHC-II expression on IECs in the small intestine and reduced donor alloreactive CD4+ T-cell expansion and Th1 differentiation, resulting in highly attenuated GVHD. Mechanistically, recipient macrophages in the ileum respond to the microbiome to secrete IL-12, which controls IFN-γ secretion in the ileum by both innate cells (principally type-1 innate lymphoid cells) and conventional T cells, the latter in particular after TBI. This information highlights the ability of gut microbiome–derived PAMP signals to invoke APC function by IECs. These local effects of PAMP signals may explain why acute GVHD target organs reproducibly include the GI tract, skin, and liver. Thus, local tissue microbiota-generated PAMP signals to both hematopoietic and nonhematopoietic APCs in the skin and GI tract and subsequently the liver via portal circulation. This pathway may thus initiate a tissue-localized cascade of donor T-cell expansion, and the study of tissue resident T-cell responses, both at the level of priming and effector phases, now deserves further study. In addition, the development of parenchyma-sparing conditioning using antibody-based approaches that target hematopoietic and immune components with71 or without radioisotope conjugates72,73 may obviate the nonspecific inflammation seen after current conditioning protocols.
It is important to keep in mind that GVHD is largely eliminated in germ-free mice,74 and dysbiosis transfers hyperacute GVHD phenotypes to mice.75 This suggests an overall stimulatory effect of microbiota in the initiation of GVHD and is congruent with clinical studies, including a phase 3 randomized trial, which demonstrated a protective effect of the intensity of antibiotic-mediated intestinal decontamination on GVHD.76,77 Indeed, successful decontamination of the intestinal microbiome in clinical recipients early after BMT is associated with the prevention of acute GVHD.78 These findings are consistent with the attenuation of GVHD in germ-free mice first noted by van Bekkum et al79 in the 1970s, and the reductions in clinical GVHD afforded by a protective environment subsequently described in the 1980s by Storb and Thomas.80 In the last decade, multiple species of microbiota, classified primarily on the basis of 16s ribosomal RNA sequencing, have been associated with GVHD, both positively and negatively. Butyrate-producing Clostridia are associated with the maintenance of epithelial barrier function and attenuation of acute GVHD.81 Intriguingly, these same butyrate-producing bacteria that are associated with reduced levels of GVHD when they are present early peritransplantation have recently been associated with steroid-refractory acute GVHD when present at diagnosis.82 In contrast, defects in Paneth cell–derived α-defensins predict systemic invasion of Escherichia coli after BMT,83,84 and some antibiotics (eg, imipenem/cilastatin) induce defects in the colon mucus barrier and are associated with increased GVHD.85 The nature of the bacterial, viral, and/or fungal species, and molecules involved in these spectra of effects, remains to be elucidated, and it is now important to understand both pathogenic and protective components of the microbiome and delineate true cause-and-effect relationships with GVHD.86,87 Of note, IECs in the ileum can constitutively express MHC-II in mice and humans,38,88 presumably depending on the nature of commensal microbiota. Likewise, the nature of costimulatory signals delivered by nonhematopoietic APCs or the neighboring cells that are required to stimulate donor T cells within tissue remains to be elucidated.
Roles of APCs in cGVHD
The risk factors for clinical cGVHD include HLA disparity, sex mismatch (female to male), and prior acute GVHD. Given this, it would seem likely that alloantigen presentation and recognition are also central to the pathophysiology of cGVHD.5 cGVHD is characterized by aberrant germinal center B-cell expansion and Th17/Tc17 and Tfh differentiation with impaired Treg recovery. Nonetheless, (donor) autoreactive CD4+ T cells that develop in the thymus of recipients lacking MHC-II on donor-derived APCs can induce cGVHD in secondary recipients, which can be prevented by thymectomy before alloSCT.27,89 The JAK1/2 inhibitor ruxolitinib is now being investigated for the treatment of cGVHD and acts to limit T-cell proliferation and Th1/Th17 differentiation via STAT inhibition.90 Interestingly, preclinical studies have shown that JAK1 inhibition may directly decrease DC antigen presentation capacity.91,92
The prolonged CD4+ T-cell lymphopenia and relative Treg deficiency characteristic of cGVHD93 can be partially reversed with low-dose IL-2 therapy, ameliorating cGVHD in a subset of patients.94,95 FoxP3+ regulatory T-cell homeostasis also requires MHC-II–dependent antigen presentation in the periphery.96 Importantly, acute GVHD, a major risk factor for cGVHD, grossly impairs the ability of donor myeloid (CD8−) DCs97 to present both donor- and host-derived antigen on MHC-II, which impairs Treg survival in the periphery. This Treg defect is causally related to the development of cGVHD.96 Recently, the expansion of donor CD11b+ DCs via GM-CSF administration has been shown to improve subsequent (FoxP3+) Treg homeostasis in the periphery and attenuate experimental cGVHD.98 Thus, the effects of GM-CSF early after BMT in acute GVHD and late after BMT in chronic GVHD are opposing.
Macrophages and B cells have important roles in cGVHD, and donor T-cell–derived IL-17 drives CSF-1–dependent donor macrophage infiltration in target tissue, which in turn mediates fibrosis.99,100 B cells from patients with cGVHD have enhanced capacity for proliferation, costimulation, and alloantibody production.101-103 Although these cell types are professional hematopoietic APCs, their roles in cGVHD have been primarily attributed to effector function rather than antigen presentation to date, such that the latter requires further study.
Antigen presentation requirements for GVL effects
Both CD4+ T cell– and CD8+ T cell–dependent GVL effects require host hematopoietic APCs and alloantigen expression on leukemic cells.10,50 Donor APCs are minimally required,50 and this is supported by studies of donor lymphocyte infusion in mixed vs full BM chimera, whereby host hematopoietic cells were required for GVL effects.104,105 Thus, in full donor chimeras (where all hematopoietic APCs are donor) in MHC-matched allograft systems, the GVL effect is abrogated.10 The GVL effects mediated by donor CD4+ and CD8+ T cells require MHC-II and MHC-I expression on leukemia cells, respectively, suggesting an absolute requirement of cognate interaction with leukemia cells.106,107 This indicates that recipient hematopoietic APCs prime donor T cells by presenting endogenous (cytosolic) alloantigen to initiate alloreactive T-cell responses, which in turn recognize alloantigen expressed by recipient leukemia. It is important to note that as for GVHD, recipient (CD8+) DCs potently delete alloreactive donor CD8+ T cells and impair GVL in experimental systems,33 and so the recipient hematopoietic APCs important in initiating MHC-I–dependent GVL remain to be determined. In contrast to the requirement for recipient hematopoietic APCs, alloantigen presentation by recipient nonhematopoietic APCs may in fact impair GVL over time.56 Thus, chronic antigen exposure by these cells promotes apoptosis and exhaustion (eg, PD-1 expression) on donor CD8+ T cells. Likewise, the ongoing presentation of alloantigen by the epithelium, in the context of PD-L1 expression in response to IFN-γ secretion from alloreactive T cells, is a potent mechanism that regulates the development of alloreactivity (and idiopathic pneumonia syndrome) after transplantation.108 Thus, chronic antigen presentation by nonhematopoietic cells seems to play an inhibitory role in both GVHD and GVL and in fact is a target for checkpoint inhibition.56
Although GVHD is primarily induced by naïve donor T cells, memory CD4+ or CD8+ T cells have been demonstrated to be capable of mediating GVL in experimental systems, albeit usually of inferior magnitude to that mediated by naïve donor T cells.107,109-111 Because memory T cells at steady state have likely been generated in response to commensal microbiota or food in mice, and additionally pathogens in humans, their response to alloantigen likely relies on heterologous immunity. The reason why memory T cells can mediate a GVL effect but not GVHD is incompletely understood; however, it seems to relate to their ability to mediate cytolytic effects necessary for GVL but their inability to undergo the full expansion or effector differentiation required for GVHD.112 The latter defect in memory T cells seems to involve enhanced AICD and exhaustion/anergy.113,114 It is therefore intriguing to consider that this may reflect a differential sensitivity of memory T cells to recipient DC–invoked AICD, and this should be further investigated. In humans, mHA-reactive CD8 T cells exist with high frequency, primarily in the naïve T-cell subset.115 Studies to date with (CD45RA+) naïve T-cell–depleted grafts116,117 have shown similar (albeit less severe) rates of acute GVHD but very low rates of chronic GVHD relative to historical cohorts. Relapse rates are not obviously increased in these transplantation cohorts undergoing intensive myeloablative conditioning, but randomized studies will be required to yield definitive data in regard to the GVL capacity of memory T cells in humans. In a majority of leukemia patients, T cells reside in BM, and the role of preferential T-cell priming and subsequent exhaustion at this site in response to malignant (and nonmalignant hematopoietic) APCs is unclear. Donor T cells can enter the BM under the guidance of selectins (eg, PSGL-1) and chemokines (eg, CXCL12, the ligand of CXCR4)118 and can respond locally to BM APCs.119,120 Certainly malignancies such as myeloma can dramatically enhance T-cell exhaustion in marrow specifically over time, even in the absence of alloreactivity,121 and so the role of antigen presentation by the primary malignancy in this process requires further investigation.
Relapse of acute leukemia after clinical haploidentical alloSCT is often associated with the loss of recipient MHC-II expression on leukemia cells, while MHC-I expression remains intact.122 Likewise, relapse after MHC-matched clinical alloSCT is associated with downregulation of MHC-II but not MHC-I on leukema.123 These studies suggest the importance of functional MHC-II by leukemic cells, either in the stimulation of leukemia-specific T-cell responses or, more likely, as a requirement for susceptibility to donor CD4 T-cell–dependent killing. Intriguingly, the low MHC-II expression on relapsed leukemia can be restored by IFN-γ,123 and this pathway has been exploited in preclinical systems to enhance the potency of GVL effects.124
Together, data to date suggest that an optimal GVL response independent of GVHD requires the stimulation of a donor T cell by recipient hematopoietic APCs presenting hematopoietic or leukemia-specific antigens, including within MHC-II. Importantly, the generation of a T-cell effector response should ideally occur in the absence of active GVHD that promotes T-cell exhaustion secondary to broad upregulation of checkpoint inhibitor molecules. The latter is particularly problematic when the T cell is directed to alloantigen expressed by both hematopoietic and nonhematopoietic targets.
Inhibition of alloantigen presentation in the gut
In conclusion, we now understand that GVHD is a complex process initiated by the synergistic interaction of microbiome and conditioning-induced DAMP/PAMP signals to recipient hematopoietic and nonhematopoietic APCs in the GI tract. Donor T-cell activation, expansion, and differentiation can occur in both lymphoid organs in response to professional APCs and tissue sites in response to nonhematopoietic APCs. Furthermore, donor T-cell–derived IFN-γ augments MHC expression and presentation at epithelial and mesenchymal surfaces. The importance of IL-12 in driving this process125 provides an obvious target for therapeutic inhibition in the early peritransplantation period, commencing before conditioning. Similarly, the microbiome is clearly important in promoting alloantigen presentation and GVHD, but subsets of bacteria also play a role in inhibiting disease. Understanding the microbes mediating these differential effects will be critical to their effective therapeutic modulation. Other pathways controlling the feed-forward cascade of GVHD in the GI tract are attractive therapeutic targets to prevent GVHD (eg, GM-CSF, IFN-γ, IL-6, TLRs, α4β7),11 but such targeting would seem unlikely to be effective once GVHD has been initiated (ie, as treatment). Finally, given the relative importance of antigen presentation by hematopoietic vs nonhematopoietic APCs in promoting GVL vs GVHD, understanding the differential pathways of antigen presentation by these cells is critical for the development of agents that will selectively inhibit APCs in tissue rather than lymphoid organs.
Acknowledgments
The authors thank Madeleine Flynn of QIMR Berghofer and James Woolace of Fred Hutchinson Cancer Research Center for generation of the graphics.
This work was supported by a research grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL148164). G.R.H. is an Andy Hill Cancer Research Endowment (CARE) Distinguished Researcher.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: M.K. and G.R.H. wrote the manuscript.
Conflict-of-interest disclosure: M.K. and G.R.H. have submitted a patent application on methods to prevent antigen presentation in the GI tract. G.R.H. has received funding from Roche for a clinical study of tocilizumab in acute GVHD prophylaxis.
Correspondence: Geoffrey R. Hill, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA, 98109; e-mail: grhill@fredhutch.org.
