

Key Points
The ThromboGenomics HTS test validates recent gene discoveries and detects CNVs and intronic variants.
ThromboGenomics reveals a molecular diagnosis for 37.3% of 2396 patients with BTPDs.

Abstract
A targeted high-throughput sequencing (HTS) panel test for clinical diagnostics requires careful consideration of the inclusion of appropriate diagnostic-grade genes, the ability to detect multiple types of genomic variation with high levels of analytic sensitivity and reproducibility, and variant interpretation by a multidisciplinary team (MDT) in the context of the clinical phenotype. We have sequenced 2396 index patients using the ThromboGenomics HTS panel test of diagnostic-grade genes known to harbor variants associated with rare bleeding, thrombotic, or platelet disorders (BTPDs). The molecular diagnostic rate was determined by the clinical phenotype, with an overall rate of 49.2% for all thrombotic, coagulation, platelet count, and function disorder patients and a rate of 3.2% for patients with unexplained bleeding disorders characterized by normal hemostasis test results. The MDT classified 745 unique variants, including copy number variants (CNVs) and intronic variants, as pathogenic, likely pathogenic, or variants of uncertain significance. Half of these variants (50.9%) are novel and 41 unique variants were identified in 7 genes recently found to be implicated in BTPDs. Inspection of canonical hemostasis pathways identified 29 patients with evidence of oligogenic inheritance. A molecular diagnosis has been reported for 894 index patients providing evidence that introducing an HTS genetic test is a valuable addition to laboratory diagnostics in patients with a high likelihood of having an inherited BTPD.
Introduction
Inherited bleeding, thrombotic, and platelet disorders (BTPDs) are a heterogeneous group of rare disorders caused by DNA variants in a large number of loci. The most common bleeding disorders are von Willebrand disease, affecting up to 0.01% of the population, and hemophilia A and B, which together affect 0.01% of the male population.1 There are no accurate estimates of the prevalence of the remaining rare inherited bleeding disorders, although registry data suggest the prevalence being <0.001%.2 Venous thrombosis has an overall annual incidence of <1 in 1000, but it is rare in the pediatric population, with rates of ∼1 in 100 000, indicative of possible environmental and lifestyle effects in adult patients.3 To obtain a conclusive molecular diagnosis requires attendance at multiple outpatient consultations for a large portion of patients with an assumed diagnosis of a rare inherited BTPD.
The genetic architecture of inherited BTPDs is well determined, but new genes continue to be identified. To date, there are close to 100 diagnostic-grade genes (hereafter TIER1 genes) associated with coagulation, thrombotic, and platelet disorders. Since validation of the ThromboGenomics high-throughput sequencing (HTS) test,4 33 TIER1 genes, including recently discovered BTPD genes, have been added to the HTS test, increasing the clinical utility. Others have reported on similar gene panel tests or used whole-exome sequencing to identify DNA variants causing inherited BTPDs.5-10 Most studies used patients with uniform clinical phenotypes, but all on relatively small numbers of patients (<160 index patients), preventing firm conclusions about the clinical utility of such tests. The diagnostic rates obtained in these studies cannot be compared as all focused on different sets of genes (with a subset of TIER1 genes, and also inclusion of “research” genes), used different patient-inclusion criteria, and variant classification was not standardized as is now recommended.11
Here, we report on the results obtained with the ThromboGenomics HTS test for 2396 index patients categorized into 5 classes of disorders based on the appended human phenotype ontology (HPO) terms12 and referral information: thrombotic, platelet count, platelet function, coagulation, and unexplained bleeding. Variant interpretation, by a multidisciplinary team (MDT), determined the contribution of variants in TIER1 genes to the observed phenotypes thereby providing insights in the clinical utility of HTS testing for different categories of patients. We also comment on the standardization of variant interpretation and how the reporting of a conclusive molecular diagnosis has immediately impacted on clinical management. Finally, due to the large number of patients tested, we are able to highlight the clinical importance of detecting copy number variants (CNVs), deep intronic variants, and possible oligogenic inheritance.
Methods
Patients
The 2396 index patients were either referrals for the ThromboGenomics test or patients who joined the PANE and Vienna Bleeding Biobank (VIBB) studies (supplemental Table 1, available on the Blood Web site). Clinical and laboratory phenotypes were recorded using HPO terms as described.4,13 Further details of the study participants, including institutional review board or research ethics committee information are in the supplemental Information.
ThromboGenomics referrals for diagnostic testing of inherited BTPDs
Samples with clinical and laboratory phenotype information from 1608 index patients with a known or suspected diagnosis of inherited BTPD, according to criteria described (supplemental Information),4 were recruited by clinicians in 72 UK and 46 non-UK hospitals (supplemental Figure 1).
PANE: preoperative screening for mild bleeding risk
A total of 212 patients, identified through preoperative assessment of bleeding risk at Maastricht University Medical Centre, were recruited; they underwent a full hematological assessment including extensive laboratory testing for hemostasis parameters (supplemental Table 2).14,15
VIBB
A total of 599 patients referred to the hematology and hemostaseology specialist tertiary referral center in Vienna for assessment of a mild to moderate bleeding disorder were recruited and subjected to a full hematological assessment, including extensive laboratory testing for hemostasis parameters (supplemental Table 2).16
ThromboGenomics HTS test
The ThromboGenomics HTS test sample preparation and sequencing protocols are as described with minor modifications (supplemental Information).4 The content of the test has been reversioned twice since first described to include additional (mostly recently discovered) TIER1 genes. ThromboGenomics version 2 (TG.V2) and version 3 (TG.V3) include 80 and 96 genes, respectively (supplemental Table 3). TIER1 genes are curated and approved by the Scientific and Standardization Committee on Genomics in Thrombosis and Hemostasis (SSC-GinTH) of the International Society on Thrombosis and Haemostasis (ISTH) (https://www.isth.org/page/GinTH_GeneLists). For TG.V3, probes for 10 000 common single-nucleotide variants (SNVs) were included to estimate relatedness and ancestry. Automated bioinformatics analysis pipeline methods, including variant calling, are described in the supplemental Information.
Variant prioritization and interpretation
Variants were annotated and prioritized for interpretation using the analytical process as reported4 based on: the predicted effect in the curated transcript, presence in the Human Gene Mutation Database17 or in a curated set of known pathogenic variants (supplemental Information), and the minor allele frequency in the Exome Aggregation Consortium (ExAC) and Genome Aggregation (gnomAD) databases.18 On a patient-by-patient basis, DNA variants passing filtering were prioritized and interpreted by an MDT in the context of the appended HPO terms, clinical information, and family history. Reported variants were characterized as pathogenic, likely pathogenic, and variants of uncertain significance alongside a decision of the likely contribution of each variant to the patient’s phenotype. The MDT made use of the Congenica diagnostic decision support platform Sapientia (Cambridge, United Kingdom) to support the review process and record findings in the form of research reports for return to referring clinicians. For all samples sequenced using TG.V2, variant interpretation was performed according to guidelines agreed to by the members of the ThromboGenomics MDT (criteria in supplemental Table 4 and details of MDT process in supplemental Information). In 2017, the UK Association of Clinical Genomic Science published best practice guidelines for variant interpretation based on the earlier reported American College of Medical Genetics and Genomics (ACMG) guidelines.11 Sapienta software implemented ACMG guidelines from release 1.7 (January 2018), allowing rapid variant interpretation following these guidelines. This updated system was applied for variant interpretation of all samples sequenced using TG.V3. Table 1 summarizes the main differences in panel content, methods, analysis, and interpretation used for TG.V2 and TG.V3.
The ThromboGenomics TG.V2 and TG.V3 HTS test content, experimental methods, analysis methods, and variant interpretation guidelines
| TG.V2 | TG.V3 | |
|---|---|---|
| Panel content | 19 Coagulation genes | 21 Coagulation genes |
| 8 Thrombotic genes | 9 Thrombotic genes | |
| 53 Platelet genes | 66 Platelet genes | |
| HGMD Pro 2015.2 variants | HGMD Pro 2016.4 variants | |
| 10 000 SNVs | ||
| Region of interest | 0.222 Mb | 0.275 Mb |
| Methods | DNA fragmentation: 220 bp | DNA fragmentation: 350 bp |
| Multiplex: 48 samples | Multiplex: 96 samples | |
| Known variants | HGMD_PRO_2017.2 | HGMD_PRO_2017.4 and curated known variants |
| Variant interpretation | MDT criteria (supplemental Table 4) | ACMG guidelines10 |
| TG.V2 | TG.V3 | |
|---|---|---|
| Panel content | 19 Coagulation genes | 21 Coagulation genes |
| 8 Thrombotic genes | 9 Thrombotic genes | |
| 53 Platelet genes | 66 Platelet genes | |
| HGMD Pro 2015.2 variants | HGMD Pro 2016.4 variants | |
| 10 000 SNVs | ||
| Region of interest | 0.222 Mb | 0.275 Mb |
| Methods | DNA fragmentation: 220 bp | DNA fragmentation: 350 bp |
| Multiplex: 48 samples | Multiplex: 96 samples | |
| Known variants | HGMD_PRO_2017.2 | HGMD_PRO_2017.4 and curated known variants |
| Variant interpretation | MDT criteria (supplemental Table 4) | ACMG guidelines10 |
Results
Patient-inclusion criteria and phenotypes
A total of 2396 index patients and 156 samples from relatives and carriers were tested using the ThromboGenomics HTS test (supplemental Table 1). The largest group of 1608 index patients and 18 referrals for hemophilia carrier status were referred by specialist tertiary centers for diagnostic testing. To better appreciate the clinical utility of the HTS test, we also included 193 and 595 index patients from the PANE and VIBB single-center studies.
Based on the clinical and laboratory phenotypes of all patients, a total of 7341 HPO terms were appended. HPO terms and clinical information were used to categorize all patients into 5 broad disease classes: thrombotic (n = 284), platelet count (n = 335), platelet function (n = 430), coagulation (n = 728), and unexplained bleeding (n = 619) (Figure 1A). The ThromboGenomics referrals included patients of all 5 classes, whereas the majority of the PANE patients (80.3%) and VIBB patients (59.5%) are classed as unexplained bleeding.
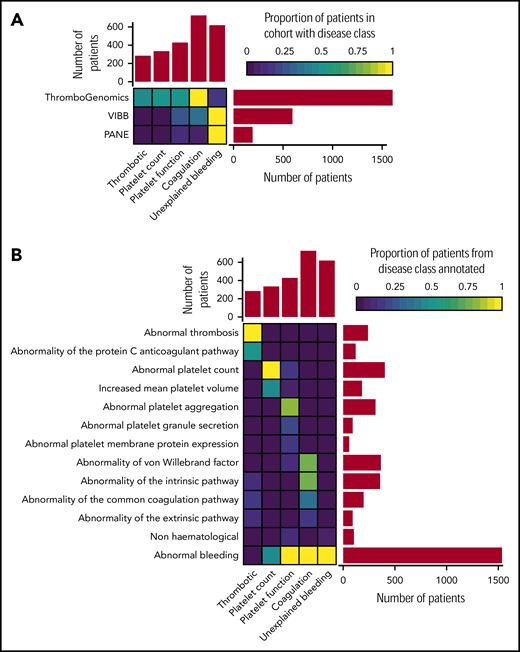
Classification of patients using clinical and laboratory phenotypes. (A) Classification of 2396 index patients from the ThromboGenomics, VIBB, and PANE cohorts into 1 of 5 disease classes: thrombotic, platelet count, platelet function, coagulation, and unexplained bleeding. (B) Representative HPO codes for patients characterized in each of the 5 disease classes.
Classification of patients using clinical and laboratory phenotypes. (A) Classification of 2396 index patients from the ThromboGenomics, VIBB, and PANE cohorts into 1 of 5 disease classes: thrombotic, platelet count, platelet function, coagulation, and unexplained bleeding. (B) Representative HPO codes for patients characterized in each of the 5 disease classes.
Most of the patients categorized to the thrombotic class were referred because of reduced protein C or protein S levels as indicated by the HPO term “Abnormality of the protein C anticoagulation pathway” (Figure 1B). Patients with platelet count disorders were generally referred due to (macro)thrombocytopenia. Platelet function abnormalities were diverse, including defects in aggregation, reduced platelet membrane protein expression (particularly glycoprotein Ib [GPIb]/IX/V and GPIIb/IIIa), and reduced granule secretion. Coagulation defects included patients with reduced von Willebrand factor levels or abnormal coagulation parameters of the intrinsic, extrinsic, and common pathways. HPO terms coding abnormal phenotypes outside of the blood system were appended to 41 of patients (1.7%). These terms related to eyes (ocular albinism), hearing (deafness), skeletal (abnormal radius, joint disorders), and kidney (functional insufficiency). Across the 3 study groups, 619 patients with bleeding symptoms and normal hemostasis test results were classed as unexplained bleeding (Figure 1B). Over half of the patients (51.2%) in this class were characterized by the presence of both spontaneous and trauma-related bleeding symptoms, a similar distribution as found for the coagulation and platelet function disease class (supplemental Figure 2). Chronic mild thrombocytopenia is generally not accompanied by spontaneous bleeding.19 However, 65.1% of the patients in the platelet count class presented with spontaneous bleeding, strongly suggesting an enrichment of “thrombocytopenia with bleeding” patients.
Performance of the ThromboGenomics HTS test
The previously reported validation of the ThromboGenomics HTS test (TG.V1) used 296 samples from patients, with and without previously known disease-causing variants, sequenced for 63 TIER1 genes.4 Here, we sequenced 1333 and 1063 index patient samples with the TG.V2 and TG.V3 tests targeting 80 and 96 TIER1 genes, with a region of interest of 0.222 Mb and 0.275 Mb, respectively (Table 1). Since validation, the analysis method for calling SNVs and short (<50 bp) insertion/deletions has undergone minor modifications; however, the detection of CNVs has been substantially improved.20 In short, sequencing read depth is computed over 500-bp elements, to improve the sensitivity for the detection of shorter CNVs, and an optimized reference set of data obtained from 10 samples has been generated, using genetically unrelated individuals (supplemental Information). Despite the increase of the region of interest and increased multiplexing of samples, the read coverage has remained high, with 99.99% and 99.98% of the region of interest with a 30× read depth for TG.V2 and TG.V3 tests, respectively. For each sample, an average of 146.7 and 190.9 SNVs, 9.4 and 11.2 insertion/deletions indels, and 0.20 and 0.21 CNVs were identified by the TG.V2 and TG.V3 tests, respectively. The proportions of variant types identified did not differ between test versions (supplemental Figure 3). For all samples, an average of 4.5 variants were prioritized for interpretation by the MDT (supplemental Table 1).
Diagnostic rates and validation of recently discovered TIER1 genes
Prioritized variants were reviewed by the MDT in the context of the disease incidence, variant frequency, assigned HPO terms, and family history. Variants were reported with pathogenicity and the contribution to the patient’s phenotype (full or partial). Variants for recessive BTPDs were reported if present in the homozygous or compound heterozygous states. Variants of uncertain significance were reported if the MDT predicted a future likely pathogenic status with additional evidence from cosegregation and functional studies. Screening of 2396 index patients resulted in an overall molecular diagnostic rate of 37.3% by reporting a total of 1031 variants in 894 index patients (Figure 2A; supplemental Table 5). Most reported variants (81.9%) were rare (<0.01%) or absent in gnomAD (supplemental Figure 4). There was a marked difference in diagnostic yield between the 5 classes: thrombotic, 48.9%; platelet count, 47.8%; platelet function, 26.1%; coagulation disorders, 63.6%; and unexplained bleeding, 3.2% (Figure 2A). For patients with thrombotic and coagulation disorders, 68.7% and 70.6% of reported variants were known variants that had previously been associated with disease, whereas for the patients with platelet count and function disorders this proportion was lower: 44.4% and 28.4%, respectively (Figure 2B). We reason that this difference reflects the fact that cataloging of pathogenic variants for coagulation disorders (especially von Willebrand disease and hemophilia A and B) began over 3 decades ago, whereas the majority of TIER1 genes for platelet disorders have only been identified over the past decade. Of the 335 patients within the platelet count class, 29 were referred under a working diagnosis of “immune thrombocytopenia refractory to treatment.” In 7 of these patients, variants were reported in genes known to be associated with thrombocytopenia (ANKRD26, ETV6, ITGA2B, TUBB1).
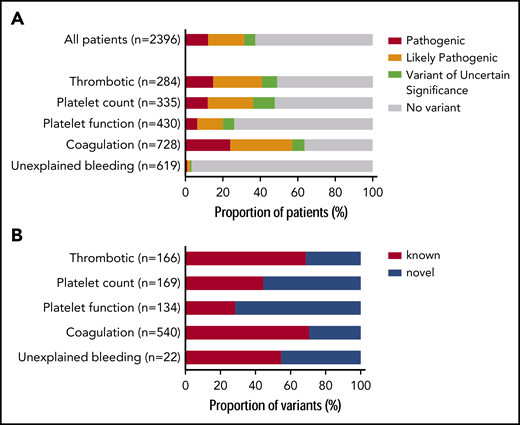
Diagnostic yield and proportion of novel variants by disease class. (A) Diagnostic yield of reported variants for 2396 index patients for each of the 5 disease classes: thrombotic, platelet count, platelet function, coagulation, and unexplained bleeding. For patients with >1 reported variant, the most pathogenic variant was used in this analysis (n = number of index patients). (B) Proportion of reported variants that were novel or known for patients in each disease class (n = number of variants).
Diagnostic yield and proportion of novel variants by disease class. (A) Diagnostic yield of reported variants for 2396 index patients for each of the 5 disease classes: thrombotic, platelet count, platelet function, coagulation, and unexplained bleeding. For patients with >1 reported variant, the most pathogenic variant was used in this analysis (n = number of index patients). (B) Proportion of reported variants that were novel or known for patients in each disease class (n = number of variants).
The implementation of the criteria of the ACMG guidelines, instead of our “in-house” criteria (supplemental Table 4), impacted on variant interpretation for patients tested using TG.V3. On comparing the ThromboGenomics cohort samples sequenced and analyzed using either TG.V2 or TG.V3 (PANE and VIBB samples were sequenced using only TG.V2 and TG.V3, respectively), there was minimal difference in the total number of reported variants for each of the 5 classes of patients (supplemental Figure 5A). However, for all disease classes, an interpretation shift from likely pathogenic to variant of uncertain significance was noted (supplemental Figure 5B). This change in variant interpretation was mainly explained by novel missense variants (supplemental Figure 6). For patients tested using TG.V2, variants that were not known as disease associated were often deemed as likely pathogenic by the MDT. On introduction of the ACMG guidelines, these novel missense variants did not reach the threshold for designation as likely pathogenic and thus were labeled as variants of uncertain significance. Prior to submission, variants were reanalyzed according to ACMG guidelines, using the latest versions of variant databases, including the ThromboGenomics database and results from cosegregation studies (supplemental Table 5, column “Reinterpretation 2019”).
The genes with reported variants in index patients are ranked in Figure 3. For the thrombotic, platelet count, and coagulation disease classes, one-quarter of patients had variants reported in just 1 gene: PROS1, MYH9 and VWF, respectively. The fourth quarter of patients for each disease class had reported variants in at least 6 genes. All reported variants per patient are summarized in supplemental Table 5 and have been submitted to the ClinVar database.21
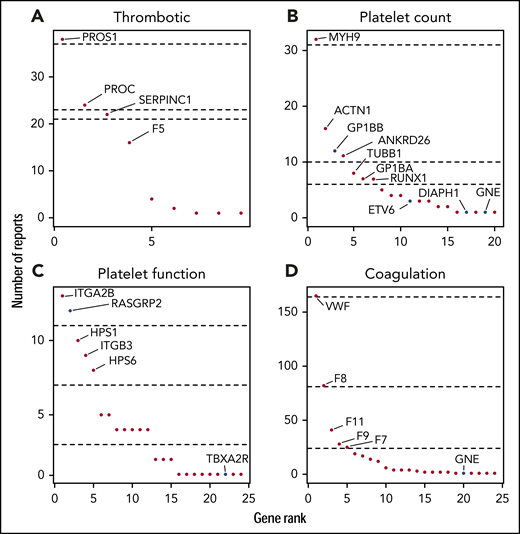
Gene ranking according to the number of reports per disease class. For each disease class, genes were ranked according to number of times they were reported (A-D). Dashed lines represents, from top to bottom, the 25th, 50th, and 75th quantiles. Recently discovered genes and changes of mode of inheritance are in blue.
Gene ranking according to the number of reports per disease class. For each disease class, genes were ranked according to number of times they were reported (A-D). Dashed lines represents, from top to bottom, the 25th, 50th, and 75th quantiles. Recently discovered genes and changes of mode of inheritance are in blue.
Since TG.V1, 17 TIER1 genes associated with BTPD have been included in TG.V2 and a further 16 more TIER1 genes in TG.V3. In addition to introducing known and recently discovered genes, those with new modes of inheritance were added (supplemental Table 3). Diagnostic reports for 41 patients have been issued with variants in 1 of the 19 recently discovered TIER1 genes or by applying a new mode of inheritance.
Copy-number variation, deep intronic variants, and oligogenic findings
The analytical pipeline for the identification of CNVs has been modified with improved quality scoring and visualization tools. Overall, CNVs were reported in 40 patients, predicted to affect single exons (n = 11), multiple exons (n = 15), or whole genes (n = 14) (supplemental Table 5). Manual inspection of reads revealed the presence of complex CNVs, including an inversion with breakpoints in introns 26 and 27 of DIAPH1 resulting in an in-frame deletion of exon 26, and an inversion flanked by 2 deletions within F8 causing severe hemophilia A (supplemental Figure 7).22
Aside from the core dinucleotide splice sites at the 5′ and 3′ of introns, the lack of reliable prediction tools makes it difficult to determine the likely functional consequences of potential splicing altering variants. Outside of the SnpEff-annotated splice regions (8 bp), intronic variants were only prioritized if previously associated with disease. A deep intronic homozygous ITGA2B variant, identified in 1 index patient with Glanzmann thrombasthenia, was validated using platelet RNA expression studies confirming alternative splicing and the absence of normal ITGA2B transcript (supplemental Figure 8). These functional data, together with cosegregation analysis in the pedigree of the index patient, resulted in a reclassification of this ITGA2B variant from variant of uncertain significance to likely pathogenic.
Of the 894 index patients for whom variants were reported, 772 had a single reported variant whereas 122 had at least 2 reported variants. Most patients with 2 variants have a recessive disease, but for 29 patients, the reported variants were in 2 or more genes. For most of these examples, the variants identified were within first- or second-order interactors in the known canonical hemostasis pathways (supplemental Table 6). For the thrombotic and coagulation classes, we identified 11 (3.9%) and 13 (1.8%) patients with oligogenic variants, respectively (Figure 4).

Oligogenic variants in patients with thrombotic (red) and coagulation (blue) disorders. From outside to inside: track 1, amino acid numbering for thrombotic (red) and coagulation (blue) genes (lighter shade denotes untranslated regions of the 3′ of the F2 gene and the 5′ of the PROS1 gene; track 2, amino acid conservation scores; track 3, variant frequency in gnomAD (minor allele frequency normalized scale to 1/106); track 4, disease-causing (red) and questionable disease-causing (orange) Human Gene Mutation Database variants; track 5 and arcs: reported variants in the 11 thrombotic (red) and 13 coagulation (blue) patients with the arcs representing oligogenic findings.
Oligogenic variants in patients with thrombotic (red) and coagulation (blue) disorders. From outside to inside: track 1, amino acid numbering for thrombotic (red) and coagulation (blue) genes (lighter shade denotes untranslated regions of the 3′ of the F2 gene and the 5′ of the PROS1 gene; track 2, amino acid conservation scores; track 3, variant frequency in gnomAD (minor allele frequency normalized scale to 1/106); track 4, disease-causing (red) and questionable disease-causing (orange) Human Gene Mutation Database variants; track 5 and arcs: reported variants in the 11 thrombotic (red) and 13 coagulation (blue) patients with the arcs representing oligogenic findings.
Incidental findings
By sequencing a large number of patients for the TIER1 genes underlying known BTPDs, we expected to observe incidental secondary findings. In 4 female patients, not referred for hemophilia carrier testing or known to have reduced factor levels, we identified carriership of likely pathogenic variants in F8 or F9. In addition, in 2 patients we identified a heterozygous deletion of the RBM8A gene. A heterozygous RBM8A loss-of function variant (generally a deletion), if accompanied by a low-frequency noncoding regulatory variant on the alternate allele, results in thrombocytopenia with absent radius syndrome.23 These incidental secondary findings were reported to the referring clinician as they are actionable with respect to family planning. In contrast, sex chromosome aneuploidy, identified in 3 patients, was not reported in line with local best practice guidelines.
Discussion
We evaluated the performance of a targeted HTS panel test for TIER1 genes in over 2500 subjects drawn from 3 distinct groups: (1) patients with a high likelihood of having an inherited BTPD, (2) patients undergoing an extended preoperative assessment for bleeding risk, and (3) patients with a bleeding disorder of unknown etiology referred to a tertiary referral center. Using HPO coding of clinical and laboratory phenotypes alongside clinical information, patients were assigned to 1 of 5 diseases classes: thrombotic, platelet count, platelet function, coagulation, or unexplained bleeding. DNA samples were sequenced with the ThromboGenomics HTS test and prioritized variants reviewed and classified by an MDT and reported to referring clinicians. The resulting data were used to assess the effectiveness of the HTS test, analytical pipeline, and MDT variant interpretation in generating a conclusive molecular diagnosis. For 1777 patients of the thrombotic, coagulation, and platelet count or function disease class, variants were reported for half (49.2%). In contrast, variants were reported for 3.2% of the 619 index unexplained bleeding patients with normal hemostasis test results. Overall, 20.1% of reported variants were variants of uncertain significance that require additional evidence including estimation of variant odds ratios using the results from large genotyped cohort studies, functional testing, and cosegregation analysis. These data illustrate the diagnostic yield obtained when applying the ThromboGenomics test for patients with a high likelihood of having an inherited BTPD.
Screening patients with the TG.V2 and TG.V3 HTS test presented several improvements compared with the TG.V1 test. Adding more TIER1 genes on TG.V2 and TG.V3 supported the molecular diagnosis of 41 patients and important genotype-phenotype associations were observed for recently discovered BTPD genes such as DIAPH1,22 ETV6,24 GFI1B,25 GNE,26,27 and RASGRP228 or for the alternative mode of inheritance recently reported for GP1BB.29 In comparison with TG.V1, the detection of CNVs was optimized, resulting in the detection of CNVs in 40 patients, including previously unobserved deletions in 13 genes, indicating CNVs as an important variant class for all categories of BTPDs. In addition, 2 novel duplications, 1 novel inversion, and a complex CNV were also reported. Nevertheless, the optimized ExomeDepth method is sensitive to variable read depths, has a minimum resolution, and cannot detect inversions or predict the location of duplicated regions. The introduction of a split-read or read-pair CNV calling method, alongside the ExomeDepth analysis method, may further improve CNV detection in the future.
Previously, Sanger sequencing of most BTPD genes was performed with primer sets flanking intron/exon boundaries and, therefore, the frequency of noncoding or silent variants, aside from those disrupting the immediate splice site (<±8 bp from the exon boundary), that are associated with BTPDs is unknown. With the use of HTS, deep intronic variants have been identified and apparent silent variants in BTPD genes have been shown to alter splicing.30-33 Variants disrupting transcription-regulatory motifs located in gene promoters and enhancer regions have also been associated with BTPDs, and with the recent mapping of endothelial and blood cell–specific enhancers, it is likely that more variants located in these regions will be identified as associated with disease in the near future.34-36 Nevertheless, due to the challenges in interpretation of noncoding variants, we have only targeted and prioritized intronic and regulatory variants if previously associated with disease. Therefore, a research analysis of novel deep intronic, silent, and likely regulatory variants detected is required, and such studies are best performed using whole-genome sequencing data from well-characterized patient cohorts.
A diagnostic HTS platform requires careful selection of diagnostic-grade TIER1 genes. The decision to include a gene to the BTPD TIER1 list is made by the GinTH SSC of the ISTH. The designation of genes for diagnostic reporting involved the review of associated literature to evaluate whether a gene is associated with disease in >3 independent pedigrees with convincing cosegregation data or in <3 pedigrees, but with strong functional evidence (mouse models or cell/protein studies) in addition to cosegregation data. In the future, this task will also be coordinated by the National Institutes of Health (NIH)-supported Hemostasis/Thrombosis Clinical Domain Working Group in close partnership with the ISTH and the American Society of Hematology (ASH) expert working groups.37
Our results indicate that when screening large number of patients with a HTS test, half of unique reported variants (50.1%) are novel and following ACMG guidelines for TG.V3, the majority (76.1%) of novel unique missense variants were reported as variants of uncertain significance (supplemental Figure 6). International initiatives for sharing of sequence data generated for BTPD patients and the sequencing of the genomes or exomes of large prospective population cohorts, like the UK Biobank, the Million Veteran Program, and the 100000 Genomes Project, will lead to statistically robust approaches for the functional labeling of DNA variants, including the variants of uncertain significance reported in this study.38-40
These data provide evidence of how a molecular diagnosis influences the clinical management and counseling of patients and their close relatives. In 30 patients, variants were reported in the ANKRD26, ETV6, and RUNX1 genes leading to counseling and follow-up due to the increased risk of hematological malignancies. These findings are associated with an important ethical debate regarding predictive testing in these genes.41 It is therefore advisable to inform clinicians and patients about the presence of genes associated with malignancies in a panel and to provide the possibility of an opt-out choice. As for whole-genome or -exome sequencing clinical-testing strategies, virtual subpanels can be used to independently analyze genes only associated with BTPDs (with and without inclusion of leukemic risk genes). In 7 patients with treatment-refractory immune thrombocytopenia, evidence of rare germline variants likely causing their condition was obtained. In 6 of these cases, the identification of a variant of uncertain significance has prompted follow-up studies in the probands and their relatives to obtain additional evidence for pathogenicity. Finally, in 24 patients with thrombotic or coagulation disorders, we reported variants in 2 or more TIER1 genes. We postulate that defects in hemostasis are due to the disruption of 2 interacting proteins in the known canonical pathways. These results of a large single study, further extend the reports of possible oligogenic architecture for thrombotic and coagulation. Together, these justify further functional and genetic follow-up studies to provide patients with a molecular diagnosis and better estimates of risk to offspring.
We report the results of the largest gene panel sequencing study of patients with suspected inherited BTPDs. We included 619 index patients with an unexplained bleeding disorder characterized by normal hemostasis test results, and, as per our hypothesis, we observed a low diagnostic rate for this group. It is possible that unknown genes are responsible or that the propensity of bleeding in these patients is the result of the aggregation of a large number of small effects emanating from common variants at hundreds of loci that are modifying the overall effectiveness of the hemostasis system. Estimation of polygenic risk scores has recently been reported for several common diseases such as coronary artery disease, type 2 diabetes, and breast cancer.42-45
In patients with a known or suspected disease etiology, a molecular diagnosis was not discovered in one-half of patients (50.8%). Future versions of the ThromboGenomics panel will include additional genes as they are identified and designated TIER1 status. However, this is unlikely to significantly increase the diagnostic rate as hundreds of genes are predicted to be currently unknown. Others have shown that reanalysis of negative clinical exome sequencing data at 1 to 3 years increased the diagnostic rate by 10% due to new evidence for gene causality and candidate variants not available during the initial analysis.46 Such evidence has emerged due to the curation of disease variants, by ClinVar and the Human Gene Mutation Database, and the availability of large exome and genome-sequencing data sets (ExAC and gnomAD). To further increase diagnostic rates in the future, the sequencing of many more individuals, through large consortium projects, will provide more accurate estimates of the rareness of potential pathogenic variants,47 alongside the development of novel genotype-phenotype algorithms,48 studies of gene-environment interactions, and the building of polygenic risk scores.
In conclusion, the ThromboGenomics test is a valuable addition to the diagnostic algorithm for patients with a high likelihood of having an inherited BTPD. The results provide clinicians with a molecular diagnosis for approximately half of patients, allowing for more precise prognostication and management of disease and, with cascade testing, better informed counseling of patients and their close relatives.
Variants and pathogenicity (Reinterpretation 2019) have been deposited in ClinVar.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study makes use of data generated by the NIHR BioResource. The authors gratefully acknowledge the participation of all NIHR BioResource volunteers and thank the NIHR BioResource center and staff for their contribution. The authors thank the Locus Reference Genomic team (LRG) for their assistance in curating gene transcripts.
This work was supported by the National Institute for Health Research (NIHR) under grant number RG65966. Research in the Ouwehand laboratory received funding from the British Heart Foundation, European Commission (TrainMALTA), International Society on Thrombosis and Haemostasis, Medical Research Council, National Health Service (NHS) Blood and Transplant, and the Rosetrees Trust. The Vienna Bleeding Biobank was supported by an unrestricted grant of CSL Behring. K.D. was supported as an NHS Higher Specialist Scientist Training (HSST) trainee by Health Education England. K.F. was supported by the Research Council of the University of Leuven (Special Research Fund [BOF] KU Leuven, Belgium, OT/14/098) and by an unrestricted grant of Sobi.
Authorship
Contribution: D.D., J.S., and C.T. performed experiments; K.M., M.V., O.S., S.V.V.D., R.M., S.T., L.D., N.G., D.G., M.H., A.T., C.J.P., and E.T. analyzed data; J.G., S.H., M.W.B., N.C., S.P., S.R.-V., S.S., E.S., W.T., I.S., Y.M.C.H., and I.P., collected samples and provided clinical support; N.A.H., H.M., and S.A. provided clinical support; and K.D., W.H.O., M.A.L., A.D.M., K.G., and K.F. led the ThromboGenomics MDT, designed the study, and wrote the manuscript, which was reviewed by all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the NIHR BioResource appears in the supplemental Appendix. A full list of investigators who contributed to the generation of the data is available from https://bioresource.nihr.ac.uk/researchers/researchers/acknowledgement/.
Correspondence: Kate Downes, Department of Haematology, University of Cambridge, NHS Blood and Transplant, Long Rd, Cambridge CB20PT, United Kingdom; e-mail: kd286@cam.ac.uk.
